On May 21, 2019, the recipients of the SFARI Bridge to Independence (BTI) Award gathered at the Simons Foundation to discuss their scientific findings and plans in autism research.
Launched in 2015, the BTI Award is a program that provides autism research funding to early-career scientists who are transitioning from a mentored (e.g., postdoctoral fellowship) to an independent research position at a North American academic institution. Researchers apply and become designated as fellows, if selected, while they are still mentees. Fellows receive a letter outlining SFARI’s commitment to funding their autism research at $495,000 over three years, which is contingent upon securing a tenure-track faculty position at a U.S. or Canadian research institution within one to two years of receiving the BTI award.
The BTI Award program includes 28 fellows, all of whom are on track to becoming SFARI active grantees. 17 fellows have started their laboratories as principal investigators and are now SFARI grantees.
Sixteen BTI fellows attended the meeting, together with SFARI senior scientists and members of SFARI review board, including Moses Chao, Anne Churchland, Kamran Khodakhah, Zach Hall, Carol Mason, Huntington Willard and Mark Zylka. The goal was to create a platform for the BTI fellows to discuss their research priorities, network and speak with senior scientists about the challenges and opportunities of becoming independent investigators.
“We organized this event with the purpose of building a community and to provide an opportunity for the fellows to discuss and learn more about autism science and research life,” said Alice Luo Clayton, SFARI senior scientist who oversees the BTI program.
BTI science
A great portion of the meeting was spent discussing progress and plans of the BTI projects. Attendees gave presentations of their scientific work, which covered diverse aspects of ASD biology, spanning from genetic causes through changes at the molecular- and neural circuit-level.
ASD genetic architecture
Mutations in over 150 genes have been linked to ASD, but how these genetic changes contribute to the condition remains little understood. A key challenge of the next generation of autism scientists will be to characterize the relationship between ASD genotype(s) and phenotype(s). To address this question, Holly Stessman studies changes in the biology (e.g., growth, morphology) of isogenic human cell lines and induced pluripotent stem cells) carrying different ASD-linked genetic mutations with the goal to identify phenotypes that are specific to genetically defined ‘subtypes’ of autism. Others, like Donna Werling, investigate the genetic and molecular mechanisms that underlie sex differences in ASD prevalence, which is typically quadrupled in boys compared to girls, to identify risk factors that make boys more susceptible and mechanisms that may protect girls, all of which are currently unknown.

Another way that BTI-funded research can contribute to advancing knowledge of autism genetics is by developing novel experimental methodologies. Reza Kalhor described a new in vivo, genetic barcoding approach that he developed to track cell lineages using the evolution of induced genetic mutations, starting from the zygote that gives rise to all the other cells of an organism,1,2. This methodology can be used to map single neuron lineages in the whole brain of developing mice, which was not possible before. In autism research, this could be applied to study the evolution of ASD-linked mutations in the brain of mouse models of the disorder (e.g., Mecp2 and Cntnap2) and help to identify, in time and space, the changes that set the ASD brain onto an atypical path from the earliest stages of development.
To understand the effects of ASD-linked mutations on brain development, another BTI fellow, Yun Li, uses in vitro assays, such as stem cells and brain organoids. In her presentation, she showed that deletion of ASD risk gene MECP2 in human embryonic stem cell-derived neurons, a model for Rett syndrome, resulted in neurons with smaller somas and dendritic arbors, reduced firing and decreased protein synthesis3 — a phenotype that is consistent with microcephaly in individuals with Rett syndrome. By contrast, deletion of PTEN, a high confidence ASD gene, produced increased proliferation of neural progenitors and brain folding in a brain organoid model, reminiscent of macrocephaly-related pathologies4.
Synaptic processes
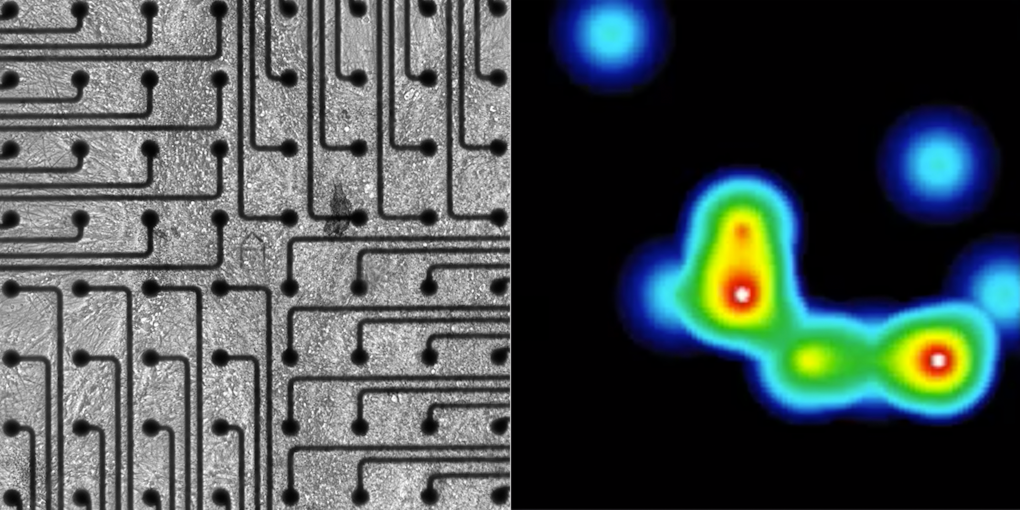
Alongside development, mutations in many ASD-linked genes are known to affect synaptic functions5. Several BTI presentations discussed the molecular mechanisms that may contribute to alterations in synaptic communication in ASD. Peng Zhang and Michael Hart focused on the role of neurexins, a family of synaptic proteins that facilitates adhesion between dendrites and axons and is encoded by genes including ASD risk genes NRXN1 and NRXN3. Zhang presented his discovery that neurexins’ synaptic functions are regulated not only by the proteins’ domain, as it was previously thought, but also by glycan modifications (e.g., through heparan sulfate, HS, proteoglycans). He showed that these molecules promote the binding between neurexin and post-synaptic ligands and mediate pre- and post-synaptic interactions, which are critical for synapse development6 (Figure 1). Hart then argued that neurexins mediate aspects of circuit plasticity; his findings in C. elegans revealed that individuals lacking nrx-1, the worm orthologue of NRXN1, exhibited reduced growth of neurites than wildtype worms, which typically grows neurites to alter circuit connectivity in response to experience7.
Another topic that was discussed was the role of glia as regulators of synaptic processes. Tingting Wang argued that glia can modulate presynaptic plasticity homeostasis, a process that helps to stabilize neurotransmitter release after perturbation8,9 and is altered in autism10. She hypothesized that genetic mutations in CHD2, an autism risk gene, may cause changes in glia and discussed her research plans to test this hypothesis in both Drosophila and mouse models lacking CHD2 in glia cells.
Also emphasizing the importance of glia/neuron mechanisms in synaptic functions, Aakanksha Singhvi then described her findings in C. elegans showing that changes in glial potassium/chloride co-transporter KCC-3 disrupted the chemical composition of the microenvironment embedding neuronal terminal endings, which, in turn, affected the neuron morphology and firing properties11. These findings not only support a role of glia in synaptic function (and dysfunction), but they also provide evidence that basic synaptic mechanisms are evolutionarily conserved and can be interrogated in invertebrate animal models. Because of its limited numbers of neurons and defined circuits, C. elegans may be a particularly useful platform to directly map molecular mechanisms to behavior, Singhvi claimed12.
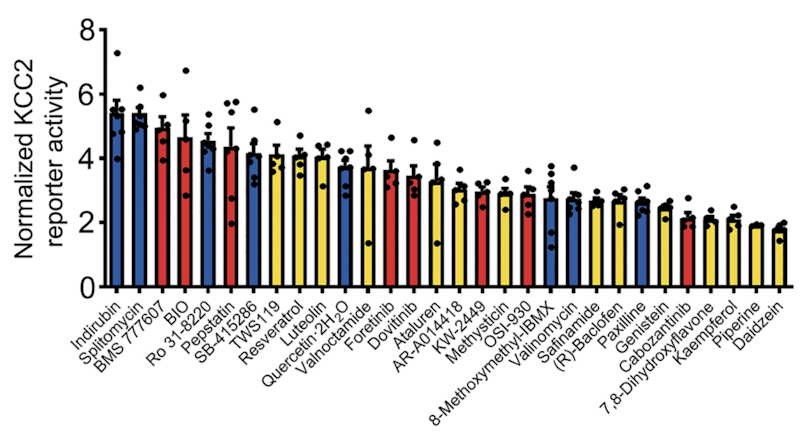
This line of research also has potential implications for developing therapeutics in ASD and related neurodevelopmental disorders where changes in synaptic transmission are observed. Xin Tang reported that the expression of potassium/chloride co-transporter KCC2 is reduced in Rett syndrome13 and argued that a treatment strategy that increases KCC2 expression levels may help to alleviate some ASD-relevant phenotypes. In support of this hypothesis, he presented data from a recent study in which he screened over 900 chemical compounds and found that a subset increased KCC2 expression and restored abnormal phenotypes to wildtype levels in human neurons lacking MECP2 and Mecp2 mutant mice, two models of Rett syndrome14 (Figure 2).
Neural circuits and systems
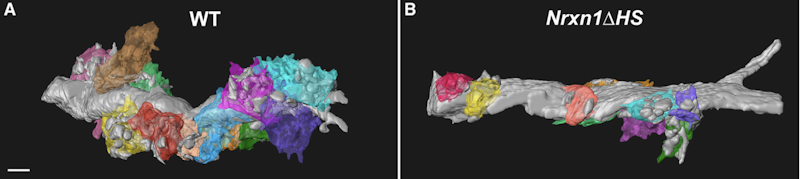
Moving from molecular mechanisms to dysfunction at the circuit level, Rui Peixoto discussed his research examining the effects of Shank3, a high confidence autism risk gene, on corticostriatal connectivity during early postnatal brain development. Mice lacking Shank3 have been previously shown to exhibit increased brain cortical activity as well as repetitive behaviors and decreased sociability; Peixoto argued that these alterations drive, as a secondary effect, earlier maturation of striatum spiny neurons and changes in corticostriatal connectivity and function15. Also highlighting the importance of early changes in the formation of neural circuits, Renata Batista-Brito then emphasized the role of genetic mutations on interneurons such as those expressing a vasoactive intestinal peptide, VIP interneurons and how alterations in these cells early in mouse brain development may affect maturation of neural circuits and behavior in adulthood16.
One way that interneurons help to regulate neural circuits is by providing inhibitory modulation via release of the neurotransmitter GABA. Changes in GABA neurotransmission have been variously implicated in ASD17, 18, 19, 20. Stephanie Rudolph, who studies cerebellar circuits in mice, argued that disruption of a specific form of GABAA receptor-mediated neurotransmission called ‘tonic inhibition’ may affect diverse behaviors, including anxiety-like and social behaviors.
She currently performs electrophysiology and behavioral testing in mice lacking the δ GABAA receptor subunit specifically in cerebellar granule cells, which receive and integrate multisensory information from many brain regions, such as the cortex, the brainstem, and the spinal cord, as well as neuromodulatory inputs that regulate tonic inhibition. Her goal is to better understand how disruption of cerebellar function may be relevant to ASD pathophysiology.
Following presentations focused on brain pathways involved in sensory processing. People with ASD often report hyper- or hypo-sensitivity to sensory stimuli, but the neural substrates of these atypical perceptions are unknown. Sun Eun ‘Samuel’ Kwon described his research in mice showing that percepts to touch are encoded at the higher levels of the ascending sensory pathway, that is, in the primary and secondary sensory cortical regions21; then, he discussed his plans to investigate intracellular mechanisms for sensory plasticity and assess their dysfunction in mouse models of ASD.
Another BTI fellow who has been studying sensory systems22, Sung Han proposed that the experience of sensory hypersensitivity in ASD may be caused by genetic mutations that impact sensory pathways for pain processing. Intriguingly, he showed preliminary results indicating that many ASD risk genes are expressed in sensory relay areas that carry painful sensations from the brainstem to the cortex.

Sociocognition
The final two presentations discussed higher-order aspects of cognition affected in ASD, with a focus on sociocogniton, the capacity to reason about and predict the behavior of others. Keren Haroush, who researches the neural bases of dynamic social interactions, described studies in which she used an experimental design inspired from the ‘prisoner dilemma’ paradigm, a hypothetical scenario employed in game theory, to study aspects of cooperation and decision-making processes23.
Gabriela Rosenblau then presented findings from her research using functional neuroimaging combined with computational modeling to investigate algorithms underlying social inferences and learning, and their neural basis and development. In her studies, which she has conducted in typically developing adolescents and adolescents with autism, she found that brain mechanisms used to predict others’ behavior vary with the age of the participants and that brain areas including the medial prefrontal and fusiform cortices mediated these differences24. She now plans to extend her research to younger children with autism, with the goal to understand “what is specifically social about the sociocognitive deficit in autism” and to translate findings into behavioral interventions.
Concluding remarks
By bringing together scientists at different stages of their careers and with diverse research backgrounds, the meeting provided a unique opportunity for the BTI fellows to discuss research programs and priorities and build the foundations for long-lasting professional collaborations. Informal conversations with SFARI scientists and SFARI review board members also provided feedback on burning questions, such as how to set up and best manage a lab, writing successful grants, how to combine research with teaching and administrative tasks and tips for maintaining work-life balance.
“This was a unique meeting. The open discussion with autism researchers from different levels, basic research to patient care, was fantastic. Being able to put my research in context of a more comprehensive narrative on autism helped me sharpen ideas and build new hypotheses to test. I also received a lot of useful advice on how to run my own lab,” said Singhvi.
“It was inspiring to share scientific interests and goals and talk openly about the challenges we all face during this stage of our careers. I received tons of advice on starting my lab from BTI fellows who recently did the same,” said Hart.
This was the first time that SFARI organized a BTI-focused meeting. As the program continues to grow, more events of this kind are likely to be held in the future.
“SFARI is very proud of each of our BTI fellows’ accomplishments to date and looks forward to witnessing their progress in their future scientific careers. I am confident that during their career each will make important contributions that increase our understanding of brain alterations that result in autism and provide novel directions for future therapeutic efforts,” said SFARI director Louis Reichardt.
References
- Kalhor R. et al. Nat. Methods 14, 195-200 (2017) PubMed
- Kalhor R. et al. Science 361, eaat9804 (2018) PubMed
- Li Y. et al. Cell Stem Cell 13, 446-458 (2013) PubMed
- Li Y. et al. Cell Stem Cell 20, 385-396 (2017) PubMed
- DeRubeis S. et al. Nature 515, 209-15 (2014) PubMed
- Zhang P. et al. Cell 174, 1450-1464 (2018) PubMed
- Hart M.P. & Hobert O. Nature 553, 165-170 (2018) PubMed
- Wang T. et al. Cell Rep. 16, 2875-2888 (2016) PubMed
- Wang T. et al. Neuron 83, 616-629 (2014) PubMed
- Tatavarty V. et al. bioRxiv (2018) Preprint
- Singhvi A. et al. Cell 165, 936-948 (2016) PubMed
- Singhvi A. & Shaham S. Annu. Rev. Neurosci. 42, 149-168 (2019) PubMed
- Tang X. et al. Proc. Natl. Acad. Sci. USA (2016) 113, 751-756 (2016) PubMed
- Tang X. et al. Sci. Transl. Med. 11, eaau0164 (2019) PubMed
- Peixoto R.T. et al. Nat. Neurosci. 19, 716-724 (2016) PubMed
- Batista-Brito R. et al. Neuron 95, 884-895 (2017) PubMed
- Rubenstein J.L. & Merzenich M.M. Genes Brain Behav. 2, 255-267 (2003) PubMed
- Gogolla N. et al. J. Neurodev. Disord. 1, 172-181 (2009) PubMed
- Fatemi S.H. et al. J. Autism Dev. Disord. 39, 223-230 (2009) PubMed
- Robertson C.E. et al. Curr. Biol. 26, 80-85 (2016) PubMed
- Kwon S.E. et al. Nat. Neurosci. 19, 1243-1249 (2016) PubMed
- Han S. et al. Cell 162, 363-374 (2015) PubMed
- Haroush K. & Williams Z.M. Cell 160, 1233-1245 PubMed
- Rosenblau G. et al. J. Neurosci. 38, 974-988 (2018) PubMed


