The Simons Foundation Autism Research Initiative (SFARI) hosted the fall 2021 virtual science meeting in a series of three webinars in October and November 2021. In total, nine SFARI-funded researchers discussed their latest findings in autism research to an audience of over 300 attendees, including current and past SFARI Investigators, SFARI collaborators and postdocs training in SFARI-funded labs. The meeting highlighted outstanding questions into the biology of autism, ranging from the role played by transcriptional programs activated during brain development, through synaptic-related mechanisms, to chromatin modifiers and mitochondrial DNA. New approaches investigating potential mechanisms linking fever to improvement in social interactions, as well as new quantitative assessments of animal motor behaviors were also discussed. Highlights of the meeting are described below.
Transcriptional regulation
A number of genes implicated in autism encode transcription factors, which kick off transcription programs that shape the development of cell types and circuits. Neural activity derived from experience can activate some transcription factors, and this interplay between experience and transcription could be perturbed in autism. Michael Greenberg of Harvard Medical School described his work on a neuron-specific molecular complex containing NPAS4, an experience-dependent transcription factor, and NuA4, a chromatin modifier. Together these form a complex with 17 other proteins, some of which have been implicated in autism or intellectual disability. Greenberg presented new, unpublished data describing the role of this complex on the development of synapses and neural activity, and a potential effect on DNA damage.
Autism risk genes also include FOXP1 and FOXP2, which encode transcription factors and are highly expressed in the striatum during prenatal development and are involved in many functions, including speech and language. In her presentation, Genevieve Konopka of the University of Texas Southwestern Medical Center detailed how FOXP1 and FOXP2 activate transcription networks that are important for setting up the different circuits within the striatum, specifically among the direct pathway and indirect pathway spiny neurons, which are critical for motor coordination and have been implicated in autism1. For example, loss of FOXP1 in indirect pathway spiny neurons in mice results in a decreased number of this cell type and also some altered motor behaviors in mice2. The remaining indirect spiny neurons show hyperexcitability mediated by decreased potassium channels3, which suggests a potential therapeutic target. Konopka is also looking at the interplay of FOXP1 and FOXP2 to understand how loss of either or both ASD risk genes in spiny neurons may affect striatal circuits and behavior.
Loss of another transcription factor, MEF2C, is associated with a monogenic form of autism. MEF2C has a hand in orchestrating activity-dependent cell and synapse development. People missing a working copy of MEF2C show autism-related features like repetitive behavior, absence of speech and hyperactivity, as well as seizures and intellectual disability4. Christopher Cowan of the Medical University of South Carolina showed that mutations associated with this syndrome interfere with DNA binding, thus uncoupling MEF2C from its action on DNA. Mice engineered to carry a similarly defective copy of Mef2c show reduced social preference, reduced ultrasonic vocalizations and repetitive jumping; these altered behaviors are mediated by excitatory neurons, inhibitory neurons and microglia5. Cowan also presented data suggesting an excess of long-range connections and a deficit of local connections in the cortex of these mice, which suggests that MEF2C orchestrates the balance of these two kinds of inputs in a given cell.
Mitochondrial and synaptic function
Many cellular mechanisms have been linked to autism, including changes in mitochondria and synapses.
Focusing on mitochondria, Zhenglong Gu of Cornell University and the Greater Bay Area Institute of Precision Medicine, Fudan University, China, presented his work examining genetic mutations in mitochondrial DNA and a new methodology he developed to sequence mitochondrial DNA at a single cell level. Mitochondria contain their own DNA but do not house the repair mechanisms that normally safeguard nuclear DNA against mutation. Gu proposed that an accumulation of mutations to mitochondrial DNA during life could contribute to a cell’s decline and alter function of the immune system, epigenetic regulation and stem cell regeneration. Findings from his work, which he has so far conducted in Huntington’s disease, show that pathogenic mitochondrial DNA mutations come to predominate with time and signal a progressive decline of mitochondrial function6. He is also working on mitochondrial DNA mutations in ASD.
Hisashi Umemori of Boston Children’s Hospital presented his efforts to understand how PCDH19, which encodes a brain-specific cell adhesion molecule, works in regulating synaptic development. Typically the PCDH19 protein is expressed at points of contact between neurons; when extracellular domains of PCDH19 on different neurons bind to each other, this stabilizes the contacts. PCDH19 molecules also bind to and cluster another cell adhesion molecule, N-cadherin, which kicks off intracellular changes important for synapse formation.
PCDH19 mutations in humans result in a disorder with seizures, cognitive difficulties and autism; though PCDH19 is located on the X chromosome, the disorder is only found in heterozygous females, not hemizygous males. Umemori presented recent experiments in mouse models that suggest an explanation for this puzzle, which involves a mismatch between PCDH19 expression across cells in the female heterozygous mice. When PCDH19 is missing on one side of a contact between neurons in the hippocampus, N-cadherin is unable to cluster appropriately, resulting in a disruption of N-cadherin signaling and mossy fiber synaptic development; when PCDH19 is missing from all cells, as it happens in the hemizygous male mice, however, N-cadherin can now bridge across contacts by themselves and cluster to kick start synapse formation7. Over-expressing N-cadherin could also rescue synaptic function in female mice with a Pcdh19 mutation, which suggests a therapeutic strategy.
Immature spine development in the brain also marks fragile X syndrome. Hye Young Lee of the University of Texas Health Science Center at San Antonio has been focusing on the role of microglia, the resident immune cells in the brain, in pruning away spines. Lee has found that microglia-specific removals of FMR1, the gene mutated in fragile X syndrome, recapitulated the increased spine density and immature spine shapes found in fragile X mouse models in both cortex and hippocampus. She is now examining these mice for autism-associated behavioral phenotypes, such as repetitive behaviors and learning and memory tasks. Experiments with microglia in culture find that those without FMR1 are more reactive in their immune responses, a property that may well inform the development of therapies attempting to restore FMR1 expression in microglia8.
Brain development
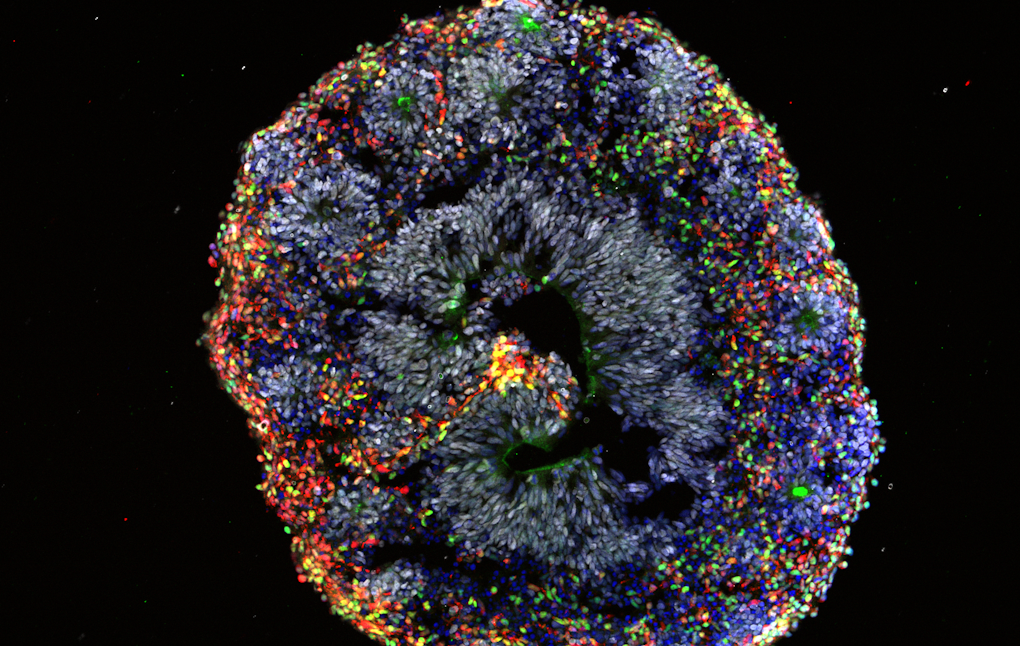
Many genes implicated in autism are active during prenatal development, which means modeling human brain development at these early stages is critical for understanding these genes’ functions. To do this, Flora Vaccarino of Yale University uses human brain organoids derived from induced pluripotent stem cells from individuals with idiopathic autism. She showed that compared to organoids derived from normocephalic individuals with autism, organoids derived from individuals with autism and macrocephaly presented a biased pattern of accelerated cell differentiation generating an imbalance of different excitatory neuron types and differences in gene expression, suggesting that head circumference may provide a useful biomarker to stratify potential subtypes of the condition. She also detailed how she and her colleagues have developed mesofluidic cell culture devices to allow the creation of morphogen gradients that provide more precise patterning of the developing organoids compared to current methods.
Motor and social behaviors
As the function of autism-related genes is explored in animal models, researchers need to optimize methods for analyzing and interpreting animal behaviors that may be relevant to autism. Bence Ölveczky of Harvard University described two methods his lab has developed to track movement at high resolution in freely behaving animals. One method affixes small retroreflective markers to the animals, allowing researchers to reliably track movements over time using a commercial motion capture system. With deep learning, this system can identify natural behavior repertoires that include rearing up, walking and grooming; it can also detect abnormalities in rat models of fragile X syndrome9. Ölveczky also described a marker-less tracking method, called DANNCE (3-Dimensional Aligned Neural Network for Computational Ethology), which uses anatomical landmarks and a convolutional neural network trained on millions of frames of video data in order to infer 3D body positions from multi-view videos10. The DANNCE system can also be used to monitor behavioral changes during development in other species like marmosets and during social interactions.
Another topic that the meeting addressed was the so-called ‘fever effect,’ according to which developing a fever may temporarily decrease hyperactivity and increase eye contact, speech and sociability in individuals with autism. Yet these changes have been anecdotally reported, and the underlying mechanisms remain unknown. Catherine Dulac of Harvard University is investigating the biological basis of this ‘fever effect’ by examining hypothalamic circuits involved in fever generation and social behaviors in mice.


