SFARI held a virtual version of its spring science meeting in a series of four webinars in April and May 2021, two of which were open to the public. The webinars were organized around themes of importance in autism research, including biological convergence, sensory sensitivity, treatment development and genetic risk. Though not the same as an in-person meeting, the online format could accommodate more attendees than usual. In the first two webinars, lab members of speakers could join, and the third and fourth webinars were open to the general public.
“In this meeting, we wanted to focus on specific topics in autism research and engage our investigators and the research community in a productive debate,” says SFARI interim director John Spiro.
Biological convergence
The first webinar explored the topic of biological convergence in autism spectrum disorder (ASD). Genetic studies have yielded over 100 risk genes for ASD, and now researchers are faced with the task of understanding the biological mechanisms that increase risk for autism and drive the many manifestations of the condition. Does each gene change need to be followed up individually to glean therapeutic insights? Or is it possible that multiple risk loci alter function in the same biological pathways, thus making it possible to generalize one therapeutic strategy to different forms of autism? In this webinar, SFARI Investigators tackled these questions from the perspectives of cell, circuit and functional networks.
To understand when and where to look for convergence, Tomasz Nowakowski of the University of California, San Francisco, seeks to create cellular-resolution maps of gene expression and chromatin accessibility in the developing human brain. A previous study highlighted the prenatal prefrontal cortex as a place where many autism risk genes are highly expressed1. Nowakowski and his team are using innovative, single-cell resolution technologies to identify the unique developmental trajectories of excitatory or inhibitory neurons in different cortical regions, including the prefrontal cortex. They have pinpointed an early molecular divergence among intermediate progenitor cells between cells destined for prefrontal cortex and those for visual cortex. Their study predicts that retinoic acid signaling promotes neural stem cells to give rise to prefrontal cortex neurons, while its absences is required by the cells to become visual cortex neurons. They validate their predictions in stem cell–based models, in which they found that modulating vitamin A levels, a precursor to retinoic acid, steered differentiation trajectories of neural stem cells into distinct molecular identifies2.
Many people with autism have touch hypersensitivity, which has led Lauren Orefice of Massachusetts General Hospital to examine the touch sensitivities in multiple mouse models of autism. Similar to their human counterparts, these mice, including those with mutations to MECP2, GABRB3 and SHANK3, show over-reactivity to touch3. Notably, selectively deleting these genes from peripheral somatosensory neurons was sufficient to reproduce this hypersensitivity, as well as anxiety-like behaviors and social impairments in the mutant mice3. This underlines the role of sensory experience on brain development, and indeed, treatments to ameliorate touch hypersensitivity in mouse models alleviated a subset of autism-related behaviors4.
John Huguenard of Stanford University presented an efficient method to assess cortical circuit function, which could help evaluate the expanding list of mouse models. It involves activating layer 5 neurons in brain slices of medial prefrontal cortex, across which an array of recording electrodes is placed. These pick up the summed neural activities that form a local field potential, which can be broken down into component parts that reflect activity at specific points of the cortical circuit. For example, brain slices made from a maternal immune activation mouse model of autism showed hypofunction among presynaptic fibers in layer 3, indicating a decrease in signals arriving to prefrontal cortex. Preliminary work in monogenic models of autism points to other types of circuit dysfunction. The assay can be completed in 10 minutes per slice allowing for a sensitive electrophysiological assay with reasonable throughput.
Jessica Cardin of Yale University and Adrian Bird of the University of Edinburgh then led a panel discussion during which participants deliberated about what convergence would look like. The variable nature of autism may mean that it encompasses several different conditions; still, like cancer, these may all involve the same kind of problem, even if the initiating causes are different. Studying different species may be a way to grasp the basic functions of circuits perturbed in autism, and there was general agreement about the importance of studies at different levels of analysis — genetic, molecular, cellular, circuit — to find convergence, and to make efforts to integrate this work across labs.
Sensory habituation
A possible point of convergence in autism is sensory habituation. People with autism often have atypical sensitivities — most frequently to auditory and touch stimuli. These can be an aversive and distressing part of their lives. One measure of sensitivity is habituation, a basic form of learning in which a repeated stimulus gradually evokes smaller responses than the first. Linda Wilbrecht of the University of California, Berkeley, who moderated the session, explained that the goal of the discussion was to seek consilience among scientists studying different species: by coming together, they might identify the experimental designs and questions that illuminate fundamental properties of brain function and genotype across species.
Catharine Rankin of the University of British Columbia studies habituation in nematode worms (C. elegans), and in her talk showed how this behavior even in a simple organism is rather complex. Her work finds that habituation itself can be broken into different components, which themselves are mediated by different genes. Investigating 135 worm strains carrying mutations in orthologs of autism-associated genes revealed some deficits in habituation5, with gene inactivation preferentially impairing response probability. She is also looking for correlations between different habituation measures to understand underlying mechanisms.
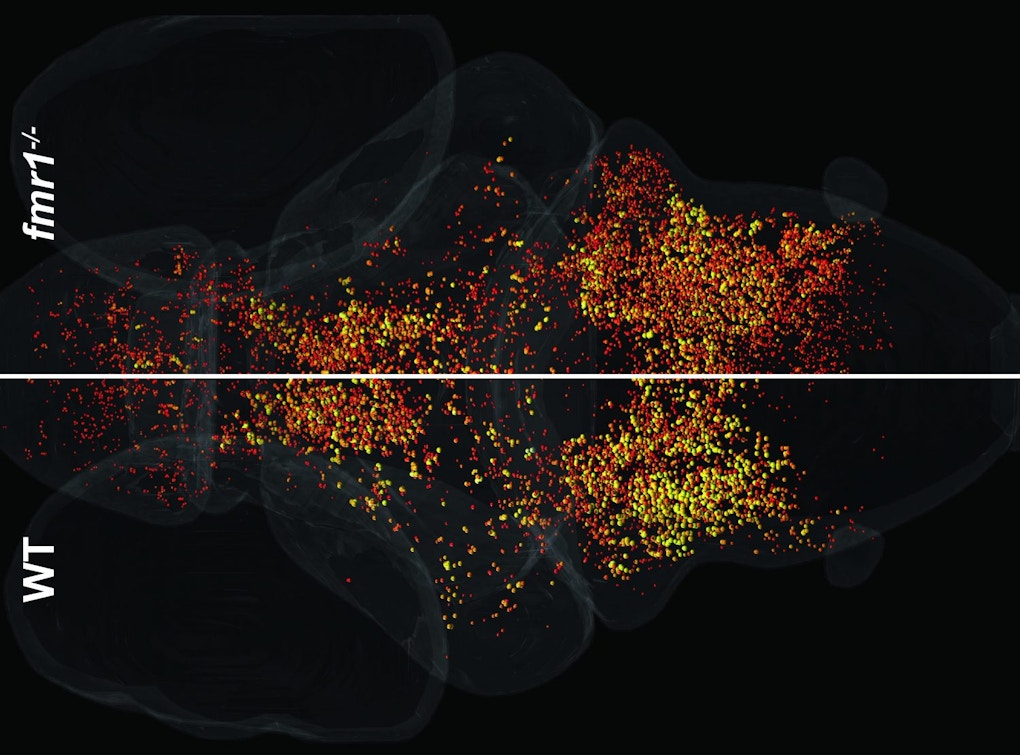
Zebrafish larvae provide a window into brain-wide responses during sensory habituation. Ethan Scott from the University of Queensland described his set-up in which zebrafish transparency, combined with whole-brain calcium imaging, allows cellular resolution of neural activity in the brain during different kinds of sensory stimulation6. One project involves responses to visual looming stimuli, which zebrafish normally swim away from. Repeated stimulation leads to habituation of response probability, as well as a mixture of neural responses7. Zebrafish lacking FMR1, the gene mutated in fragile X syndrome, are slower to habituate than controls, which may reflect a change in the network structure. Scott concluded by saying that this system is good for characterizing what happens and when it happens, but less good for answering why the changes occurred, highlighting the need for mechanistic studies to follow the initial whole-brain imaging.
Given that atypical touch sensitivity is predictive of autism severity8, Andreas Frick at INSERM has been studying touch processing in mice. He does this one neuron at a time with whole cell recordings from pyramidal neurons in layer 2/3 in somatosensory cortex. In mice lacking FMR1, he has catalogued multiple changes consistent with hyperexcitability, such as increased spontaneous firing rates, broader action potentials and larger excitatory potentials elicited by paw stimulation9. Spike timing is also affected, with increased jitter and trial-to-trial variability in the FMR1 knockout mice. The findings raise questions about how atypical sensory processing in autism may contribute to cognition.
Shulamite Green of the University of California, Los Angeles, studies habituation in people with autism, using functional magnetic resonance imaging (fMRI) to monitor brain activation. She has found that somatosensory and auditory cortex, as well as the amygdala, are hyper-responsive to auditory and touch stimulation in children and adolescents with autism, and that this correlates with their sensitivity severity10. In a habituation paradigm, children with sensory over-responsivity showed brain responses that initially decrease, then rebound — as though the brain was unable to maintain the reduced, habituated response11. Her studies also suggest that these habituation differences may stem from altered brain connectivity12. Green argued that sensory regulation and filtering may be affected in autism, so that some individuals have difficulty tamping down the brain’s response to stimulation, whereas others may be tamping it down too strongly, thus making it harder to process and respond to new information.
The panel discussion covered a number of issues. These included whether different perceptions of incoming stimuli could explain differences in habituation; how arousal state could affect sensory responses; how initial sensory responses may correlate with other metrics of habituation; finding sensory stimulation paradigms that can be standardized and translated across species, including humans; the need for quantitative and rigorous studies of habituation in people with autism; and whether there is a developmental component to habituation.
Treatment development for ASD
As basic science research continues, work on treatments for autism is already underway, in the form of small molecules, gene therapy and anti-sense nucleotides (ASOs). Federico Bolognani of Axial Therapeutics made the case for small molecules, noting that 85 percent of people with autism were idiopathic cases, in which people likely carry different combinations of risk genes, making gene therapies difficult. Many of these genes operate in the same biological pathways, and recent studies show similar patterns of connectivity in the brain in four different cohorts with autism13. Small molecules could target these converging points in the molecular pathways. The use of small molecules comes with several advantages, such as their ease of use, oral administration, adjustability of doses and reversibility of effects. One approach centers around the gut, which may also play an important role in autism: Axial Therapeutics has been working to find a small molecule that interferes with neuroactive metabolites from the gut microbiome before they are absorbed in the gut and reach the brain where they modify brain function and behavior.
Stuart Cobb of Neurogene and the University of Edinburgh said the field was on the cusp of a tsunami of gene therapies entering clinical trials for neurodevelopmental conditions. Gene therapy is appropriate for conditions caused by loss of function of a single gene, as it aims to deliver a replacement “transgene.” This approach targets the root cause of the disorder, and requires only a one-time treatment; however, it is difficult to deliver genes to the nervous system, and the genes themselves can be large and complex, with many isoforms. Proof-of-concept gene therapy trials are ongoing for Angelman syndrome, Rett syndrome and fragile X syndrome, all monogenic conditions that have features of autism.
Based on his work on Rett syndrome, Cobb listed important factors to consider when weighting a gene therapy approach: which features of the condition are reversible, the nature of the gene (how large, how many isoforms), how many cells need to be rescued genetically and the dosage. For example, too little of the protein encoded by MECP2 results in Rett syndrome, but too much is also pathologic. He also noted that delivering transgenes to the immunoprivileged nervous system does not necessarily avoid immune-related adverse reactions, as the virus vector eventually finds its way to the rest of the body.
Antisense oligonucleotides (ASOs) can also be used to regulate function of existing genes, as discussed by Yael Weiss of Ultragenyx. Typically, ASOs bind to mRNA to halt its translation into a protein, but they can also be used to upregulate protein levels. The latter approach is being explored in an ASO trial for Angelman syndrome, which is caused by a loss-of-function mutation to the gene copy of UBE3A inherited from the mother. In partnership with the Foundation for Angelman Syndrome Therapeutics (FAST), Ultragenyx has been developing an ASO that can reactivate the silenced paternal gene to compensate for the damaged maternal one. Preliminary results from a small phase 1/2 study show that upon receiving this ASO, children with Angelman showed striking improvements, but adverse reactions have recently halted the trial.
Discussion moderator Randall Carpenter of the Rett Syndrome Research Trust and Allos Pharma guided the conversation, in which participants agreed that biology should guide treatment approach. For example, small molecules may have a place for some monogenic conditions without a clear characterization of the causal mutations, if a node in a biological circuit can be identified. Modulating the epigenome to regulate gene expression, as is being done in cancers, is another possibility; however, the known modulators are not very specific, and not enough is yet known about the ASD epigenome. Regarding the number of cells that need to be rescued in a gene therapy or ASO approach, there may be a gradient of effect rather than a threshold, as has been suggested by gene therapies for certain focal epilepsies.
The prospect of treatment studies in humans emphasizes the need for sensitive outcome measures for these trials. This means developing tools that can detect small changes and also understanding the typical developmental trajectory for people with these disorders. Researchers also need to survey parents for what changes would be most important to them; small improvements in a narrow domain of cognition may not be as important to them as gains in other areas.
What do we mean by “autism risk genes”?
The final webinar of the meeting centered on the relationship between autism and other neurodevelopmental disorders (NDDs). David Ledbetter of Dascena and Joseph Buxbaum of the Icahn School of Medicine at Mount Sinai discussed whether any genes identified in genetic studies of autism confer risk for autism specifically, or instead raise general risk for brain development disorders. The issue came to the fore in a landmark paper that identified 102 risk genes for autism14. An analysis in that study found some of these genes were associated more frequently with autism than with other NDDs, which raised the question of whether there is such a thing as an autism-specific gene15,16,17.
Ledbetter argued against this terminology, noting that there is not evidence for genes to be uniquely found in autism. Buxbaum agreed that clinically, the distinction between autism and other NDDs is not worth emphasizing. However, he argued that looking for genes that were mutated more often in autism than in NDD (calling these “autism-predominant” genes rather than “autism-specific”) could be helpful for research. For example, people with ASD who carried mutations in the ASD-predominant genes walked sooner, had higher IQs and showed a different inheritance pattern of mutation than those with mutations in the other genes14. ASD-predominant genes also tended to be expressed later, in more mature cortical neurons than the other genes.
During the discussion, Heather Mefford of St. Jude Children’s Research Hospital fielded questions. A recurring theme was the importance of how cohorts are ascertained and evaluated: the reality is that autism and NDD cohorts are ascertained differently and are not uniformly evaluated for autism, NDD or intellectual disability, which makes them difficult to parse. Buxbaum agreed that ideally cohorts would be uniformly genotyped and phenotyped, but that the Autism Sequencing Consortium wanted to see what they could find out with the data they had. Future cohorts of broadly defined NDDs that have been uniformly evaluated would help, and some day research-grade genotyping and phenotyping might be efficiently achieved through routine clinical care. Also, the speakers expected that whole-exome sequencing would soon replace genetic panels as a standard genetic test.
As for where common variants fit into ASD genetic risk, genome wide association studies are still amassing risk variants. But once found, these common risk variants considered together may index a background genetic risk that can modify how a rare variant is expressed, as well as help explain phenotypic variability, which has happened recently for schizophrenia18.
If there are no autism-specific genes yet, then perhaps people should also reconsider the term “autism mouse model.” The speakers agreed that mice may not the best experimental system for modeling aspects of human behavior that are relevant to autism, such as sociality, but that they could still provide valuable insights about learning, brain development and motor functions.
References
- Willsey A.J. et al. Cell 155, 997-1007 (2013) PubMed
- Ziffra R.S. et al. bioRxiv (2020) Preprint
- Orefice L.L. et al. Cell 166, 299-313 (2016) PubMed
- Orefice L.L. et al. Cell 178, 867-886 (2019) PubMed
- McDiarmid T.A. et al. Proc. Natl. Acad. Sci. USA 117, 656-667 (2020) PubMed
- Constantin L. et al. BMC Biol. 18, 125 (2020) PubMed
- Marquez-Legorreta E. et al. bioRxiv (2019) Preprint
- Hilton C.L. et al. J. Autism Dev. Disord. 40, 937-945 (2010) PubMed
- Zhang Y. et al. Nat. Neurosci. 17, 1701-1709 (2014) PubMed
- Green S.A. et al. JAMA Psychiatry 72, 778-786 (2015) PubMed
- Green S.A. et al. Am. J. Psychiatry 176, 1010-1020 (2019) PubMed
- Wood E.T. et al. Transl. Psychiatry 11, 39 (2021) PubMed
- Holiga S. et al. Sci. Transl. Med. 11, eaat9223 (2019) PubMed
- Satterstrom F.K. et al. Cell 180, 568-584 (2020) PubMed
- Myers S.M. et. al. Am. J. Hum. Genet. 106, 587-595 (2020) PubMed
- Buxbaum J.D. et al. Am. J. Hum. Genet. 107, 1000-1003 (2020) PubMed
- Myers S.M. et al. Am. J. Hum. Genet. 107, 1004 (2020) PubMed
- Davies R.W. et al. Nat. Med. 26, 1912-1918 (2020) PubMed
- From parent advocate to nonprofit chief science officer, to biotherapeutic company cofounder — A personal journey through drug development for Angelman syndrome
- SFARI fall 2020 science meeting highlights new developments in autism research
- SFARI workshop explores challenges and opportunities of gene therapies for autism spectrum disorder
- Coordinating animal- and human-based research on sensory alterations in autism spectrum disorders


