
On February 6–7, 2020, the Simons Foundation Autism Research Initiative (SFARI) convened a two-day workshop to explore the possibility of gene therapies for autism spectrum disorder (ASD), a neurodevelopmental condition associated with changes in over 100 genes. Inspired by the recent, stunning successes of gene therapy for the fatal neuromuscular disorder spinal muscular atrophy (SMA)1, and by the accumulation of genes confidently associated with ASD2, SFARI welcomed a diverse collection of researchers to begin to think about whether a similar approach could be taken for ASD. Because gene therapy attempts to fix what is ‘broken’ at the level of a causative gene, it would offer a more direct and imminent strategy than mitigation of the many — and as yet mostly unclear — downstream effects of a damaged gene.
The workshop was organized in 20 talks and several discussion panels, which tackled many outstanding issues, including how to choose candidate target genes and predict outcomes; how to optimize vectors for gene delivery; how to decide when to intervene; which animal models to develop; how to find appropriate endpoints for clinical trials and understand the available regulatory pathways. SFARI also raised the question of how its funding might best propel gene therapy efforts amid the emerging, complex ecosystem of academic laboratories, biotech companies, and pharmaceutical industries.
“Even the opportunity to have this discussion is very rewarding,” said SFARI Investigator Matthew State of the University of California, San Francisco (UCSF), one of the investigators who directed teams of geneticists to analyze the Simons Simplex Collection (SSC).
These efforts have offered up multiple potentially feasible therapeutic targets. Though rare, de novo disruptive mutations in the highest confidence ASD genes often result in severe impairment characterized not only by social difficulties, but also by intellectual disability and seizures. The combination of a single gene mutation of large effect coupled with particularly severe outcomes that include ASD are likely to offer the most immediate targets for gene therapy. For now, this leaves out a large number of individuals with autism for whom genetic causes are not yet known and are likely the result of a combination of many small effect alleles across a large number of genes.
Genomics
Highlights from talks and discussion panel, chaired by Rick Lifton of Rockefeller University
- ASD cases associated with strong effects of high-confidence single genes are a first target. These conditions are often also associated with intellectual disability, motor difficulties and seizures.
- Cases in which one allele is disabled (haploinsufficiency) are a focus for gene therapy because the direction of effect is clear (need to add back or boost activity of a remaining good copy).
- A gene therapy toolbox includes adding back a working copy of a gene, CRISPR-mediated ways of making a good gene copy work harder and antisense oligonucleotides (ASOs) to modify transcription.
- Though nonhuman primate models will likely be necessary steps for human gene therapy trials, models in intermediate species that can be developed more quickly and economically shouldn’t be overlooked.
In the first talk of the workshop, State brought the group up to speed on ASD genomics. The most recent tally from exome-sequencing in simplex cases of ASD highlighted 102 genes in which rare mutations confer individually large risks2. In contrast, the task of identifying common variants carrying very small risks remains quite challenging, with less than a half dozen alleles so far identified with confidence3. The rare, disruptive mutations that result in loss of function of one gene copy are an attractive focus for gene therapy because of the tractability of targeting a single spot in the genome per individual and because, in the vast majority of cases, there remains a single unchanged allele. This points to ways to boost gene and/or protein expression back toward the normal state by leveraging the unaffected copy. But both the limited number of cases known so far combined with the possibility that different mutations to the same gene may have different effects complicate thinking about how to prioritize targets for gene therapy.
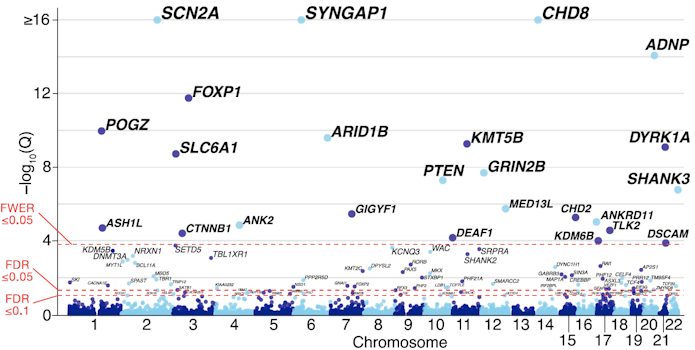
State made several points that were continually touched on throughout the workshop. Many ASD genes are highly expressed during midfetal development in the cortex, and additional experiments will need to determine whether and how long a window of opportunity may be present for successful gene therapy postnatally. Given the relatively small number of people with these conditions, new clinical trial designs are needed that don’t rely on comparisons between large control and intervention groups (see also Bryan King’s talk below).
Beyond the gene-crippling mutations found in the exome, disruptions to transcription may also dramatically raise risk for autism — and may be corrected with a type of gene therapy using ASOs. SFARI Investigator Stephan Sanders of UCSF focused on the role of splicing, the process by which an initial transcript is turned into messenger RNA by removal of introns and joining together of exons. Splicing is disrupted in at least 1.5 percent of individuals with ASD4, and possibly many more, as suggested by transcript irregularities found in postmortem autism brain5. Sanders described Illumina’s Splice AI project in which machine-learning helps predict noncoding variants that can alter splicing, including those beyond typical splice sites found near a gene6. As a result of incorporating sequence information around and between splice sites, this computational tool detected more mutations with predicted splice-altering consequences in people with ASD and intellectual disability than in those without the condition.
An ASO designed to bind specific portions of RNA could conceivably correct errors in transcription. ASOs have already been approved for use in other disorders in order to skip exons, retain exons or to degrade mRNA. Unlike other forms of gene therapy, ASOs do not permanently alter the genome, making it a kind of ‘gene therapy lite.’ This reversibility has both disadvantages (having to re-infuse the ASO every few months) and advantages (multiple opportunities to optimize the dose and target; serious adverse effects are not permanent).
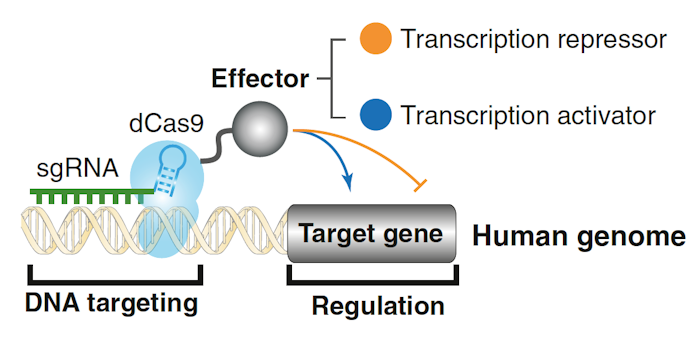
Jonathan Weissman of UCSF discussed the available toolbox for controlling gene expression developed by many different laboratories. To turn genes on or off, he has developed a method to combine CRISPR with an enzymatically inactive (‘dead’) Cas9, which can then be coupled with a transcriptional activator (CRISPRa) or repressor (CRISPRi)7 (Figure 2). In the case of loss-of-function mutations, Weissman outlined strategies to make the remaining good allele ‘work harder’: increase transcription via CRISPRa, decrease mRNA turnover, increase translation of a good transcript via modification of upstream open reading frames (uORFs) or increase a protein’s stability, possibly through small molecules acting on the ubiquitin system8. That said, the effects on a cell may be complicated. Using Perturb-Seq screens, Weissman described ‘genetic interaction manifolds’ that show nonlinear mapping between genotype and single cell transcriptional phenotypes9. Additionally, Weissman summarized recent work from his laboratory that has identified large numbers of uORFs that result in polypeptides, some of which affect cellular function.
SFARI Investigator Michael Wigler of Cold Spring Harbor Laboratories echoed the idea of a gene-therapy strategy that increases expression of the remaining good copy of a gene, especially given that in his estimate, 45 percent of simplex cases of autism carried a de novo, likely disrupting variant. He also called attention to the uterine environment, especially the challenge posed by expression of paternally derived antigens in the fetus and the impact of a potential maternal immune response, and the need to understand how it interacts with de novo genetic events.
Vectors and delivery
Highlights from talks and discussion panel, chaired by Arnon Rosenthal of Alector
- Adeno-associated virus (AAV) will need to be optimized for more efficient ability to cross the blood-brain barrier and to deliver a transgene to the brain; the LY6a gene is an important mediator of AAV’s ability to cross the blood-brain barrier in mice.
- There is an age dependence on virus uptake, with greater anatomic distribution in young than in older animals. This suggests that early interventions may have a better chance of sufficient AAV uptake in humans and would need to be tested in nonhuman primates early in development.
- Even with intrathecal administration, it is hard to get enough AAV into the brain; this highlights a need to increase the efficacy of each particle (through vector or promoter optimization) or to find ways to deliver viruses to specific brain locations.
The discussion turned to finding ways of getting genes into the central nervous system. The AAV is the darling of gene therapy, given that it does not replicate and is not known to cause disease in humans. A version that can cross the blood-brain barrier (AAV9) was used to deliver a gene replacement to children with SMA intravenously; though this effectively delivered the genetic cargo to ailing motor neurons in the spinal cord, it does not work that well at delivering genes throughout the brain.
Ben Deverman of the Stanley Center at the Broad Institute of MIT and Harvard detailed his efforts to optimize AAV for efficient transduction of brain cells through a ‘targeted evolution’ process: his team engineers millions of variants in the capsid of the virus, then screens them for entry into the nervous system and transduction of neurons and glia. This has yielded versions (called AAV-PHP.B and AAV-PHP.eB) that more efficiently enter the brain10,11. One successfully delivered the MECP2 gene to the brain of a Rett syndrome mouse model, resulting in ameliorated symptoms and an extended lifespan12. Unfortunately, these viruses don’t work in human cells or in all mouse strains. A quick mouse genome-wide association study (GWAS) revealed that the Ly6a gene mediates efficient blood-brain barrier crossing of AAV-PHP.B and AAV-PHP.eB13. Now his group has identified Ly6a-independent capsids that may translate better to humans. He also noted that the PHP.B vectors have tissue specificity for brain and liver.
With an estimated 87 percent of autism-associated genes raising risk through haploinsufficiency (having only one functional gene copy out of the two), SFARI Investigator Nadav Ahituv of UCSF made the case for approaches that boost expression of the remaining good copy of a gene through endogenous mechanisms — a strategy he called ‘cis-regulation therapy.’ This method also provides a way to work around the small four kb payload of AAV, which strains to contain cDNA of many autism genes. A recent study by his group used CRISPRa targeted at an enhancer or promoter of SIM1 and promoter of MC4R, both obesity genes, in mice. Using one AAV vector for a dCas9 joined to a transcription activator, and another AAV vector having a guide RNA targeting either a promoter or an enhancer, and a guide RNA targeting a promoter, the researchers injected the vectors together into the hypothalamus, which resulted in increased SIM1 or MC4R transcription and reversed the obesity phenotype brought on by loss of these genes14. Targeting regulatory elements had the added benefit of tissue specificity, and there seemed to be a ceiling effect for SIM1 expression, which suggested an endogenous safeguard against overexpression at work. He is now collaborating with SFARI Investigator Kevin Bender, also at UCSF, to apply this approach to the autism gene SCN2A.
Botond Roska of the Institute of Molecular and Clinical Ophthalmology in Basel, Switzerland pointed out that getting genes to the cells where they are needed is crucial when treating eye diseases. Off-target effects there can induce degeneration of healthy cells. For this reason, Roska and his group have created AAVs that target specific cell types in the retina by developing synthetic promoters that efficiently promote expression of the virus’s cargo15. The promoters they designed were educated guesses based on four approaches: likely regulatory elements close to genes expressed with cell-type specificity in the retina, conserved elements close to cell type–specific genes, binding sites for cell type–specific transcription factors and open chromatin close to cell type–specific genes. Screening a library of these in mouse, macaque and human retina revealed some with high cell-type specificity (Figure 3). Importantly, macaque data predicted success in human retina much better than did mouse data. In preliminary experiments, and more relevant to gene therapy for ASD, these cell-specific vectors also had some success in mouse cortex, for example lighting up parvalbumin neurons or an apparently new type of astrocyte.
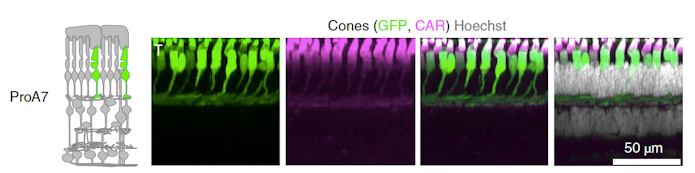
Roska also described new methods for delivery, in which nanoparticles are coated with AAV, then drawn into the brain using magnets16. This ‘magnetophoresis’ technique allows a library of experimental AAVs to be tested at the same time in one monkey. Steering nanoparticles with magnets gives more control of vector placement and gene delivery. He argued that these in the future could access even deep structures of the brain.
Current status of gene therapy for ASD-related conditions
Highlights from talks and discussion panel, chaired by Steven Hyman of the Broad Institute at MIT and Harvard
- ASO therapy may be a worthwhile intermediate step before more permanent gene replacement approaches.
- Natural history data will be important as a comparison for a treatment group in clinical trials, particularly for rare conditions. Simons Searchlight hopes to be a centralized database for this kind of data to expedite formal natural history studies, but delays in genetic diagnosis means data for the earliest stages of development may be missed.
- Intervening earlier has better outcomes in mouse models of Angelman syndrome and Phelan-McDermid syndrome. In models for SCN2A-associated changes and Rett syndrome, intervening late in development can still rescue the phenotypes tested.
- A centralized facility for nonhuman primate model development may be recommended because of the expense and expertise required for such work, an idea also endorsed by a National Academy of Sciences workshop in 201817.
- Despite extensive preclinical work in animal models, early small studies in humans often reveal critical issues not encountered in the preclinical work.
- Human models of disease such as skin-derived induced pluripotent stem cells (iPSCs) grown into neurons or brain organoids could provide useful high-throughput systems to examine mechanisms and new therapeutics.
- Of the high-confidence autism genes discovered so far, all present challenging targets for gene therapy. To prioritize them, researchers might look for those that are simpler with respect to splice variants, those that have a ceiling effect on their expression and those that act cell autonomously.
Kathy High of Spark Therapeutics reviewed the story of gene therapy for spinal muscular atrophy (SMA) type 1. Though she was not directly involved in that research, she is well aware of the regulatory atmosphere surrounding gene therapy, given that Spark Therapeutics developed the first approved AAV-delivered gene for a form of retinal dystrophy. The SMA story is a useful case study in that an ASO-based therapy (nusinersen, marketed as Spinraza), approved in 2016, set the stage for a gene-replacement therapy, marketed as Zolgensma (onasemnogene abeparvovec). Ultimately, the amount of data supporting Zolgensma’s approval was modest: a Phase one dose study of 15 infants1, and an ongoing Phase three trial of 21 infants and safety data from 44 individuals. Yet the approval was helped by the dramatic results and clear endpoints: those receiving a single intravenous infusion of an AAV9 vector containing a replacement gene all remained alive at 20 months of age, whereas only 8 percent survived to that age in the natural history data, which compiles the disease’s untreated course. High mentioned that maintaining product quality for gene therapeutics may prove trickier than for typical medications.
The attractive, highly customizable nature of gene therapy might have a regulatory downside in that different vector payloads, even when designed to do the same thing, could invite separate approval processes. Though not knowing how regulatory agencies would view this, High said that their perspectives are bound to evolve as more gene therapy trials are completed.
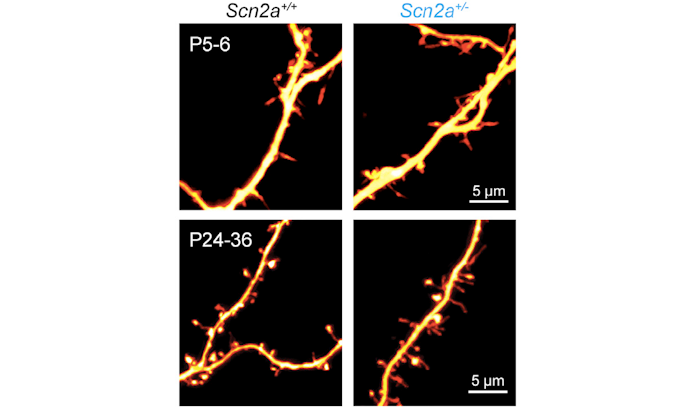
Getting to ASD-related syndromes, Bender talked about SCN2A, which encodes the sodium channel Nav1.2. SCN2A mutations in humans can be gain of function or loss of function; gain-of-function mutations are associated with early onset epilepsy, and loss-of-function mutations with intellectual disability and ASD. In a mouse model missing one copy of SCN2A, Bender and his group have discovered a role for SCN2A in action potential generation in the first week after birth, and in synaptic function and maturation afterward through regulation of dendritic excitability18 (Figure 4). Using AAV containing CRISPRa constructs developed with the Ahituv lab, the researchers successfully increased SCN2A expression, and recovered synapse function and maturity, even when done several weeks postnatally. Getting the appropriate dosage is critical since gain-of-function mutations are linked to epilepsy. However, Bender reported even when SCN2A expression increased to double normal levels, no hints of hyperexcitability appeared. “We might be able to overdrive this channel as much as we want and actually may not have risk of producing an epileptic insult,” he said. Next steps are to figure out the developmental windows for intervention, evaluate changes in seizure sensitivity and extend this kind of cis-regulatory approach to other ASD genes.
Angelman syndrome is another condition that attracts interest for gene therapy, in part because neurons already harbor an appropriate replacement gene. Angelman syndrome stems from mutations to the maternally inherited UBE3A gene, which is particularly damaging to neurons because they only express the maternal allele, while the paternal allele is silenced by an antisense transcript. SFARI Investigator Mark Zylka of the University of North Carolina and colleagues showed in 2011 that this paternal allele could be unsilenced with a cancer drug in a mouse model of Angelman syndrome19. Since then, three companies have built ASOs to do the same thing, and these are going into clinical trials. To get a more permanent therapeutic, Zylka has been developing CRISPR/Cas9 systems to reactivate paternal UBE3A, and preliminary experiments show that injecting this construct into the brains of embryonic mice, and then again at birth, results in brain-wide expression of paternal UBE3A and is long-lasting (at least 17 months). Zylka is now making human versions of these constructs. He later noted rare cases of mosaicism for the Angelman syndrome mutation — people with 10 percent normal cells in blood have a milder phenotype20, which suggests that even inefficient transduction of a gene vector could help.
Zylka also made a case for prenatal interventions in Angelman syndrome: studies of mouse models indicate that early reinstatement of UBE3A expression in mouse embryos rescues multiple Angelman syndrome-related phenotypes, whereas later postnatal interventions rescue fewer of these21; for humans, a diagnostic, cell-based, noninvasive prenatal test will be available soon22; ultrasound-guided injections into fetal brain of nonhuman primates have been developed23; prenatal surgeries are now standard of care for spinal bifida; and intervening prenatally decreases the risk of an immunogenic response to an AAV vector or its cargo. During the discussion, it was noted that another benefit of acting early was that less AAV would be needed to transduce a much smaller brain; however, a drawback is the lack of data on Angelman syndrome development from birth to one year of age. This natural history would be necessary for understanding whether a prenatal therapy is more effective than treatment of neonates.
SFARI Investigator Guoping Feng of the Massachusetts Institute of Technology has been investigating SHANK3, a high-confidence autism risk gene linked to a severe neurodevelopmental condition called Phelan-McDermid syndrome, which is marked by intellectual disability, speech impairments, as well as ASD. SHANK3 is a scaffold protein important for organizing post-synaptic machinery in neurons. Mouse studies by Feng have shown that SHANK3 re-expression in adult mice that have developed without it can remedy some, but not all, of their phenotypes, including dendritic spine densities, neural function in the striatum and social interaction24. Furthermore, early postnatal re-expression rescued most phenotypes. This makes SHANK3 a potential candidate for gene therapy; however, it is a very large gene — 5.2kb as a cDNA — that is difficult to fit into a viral vector. To get around this, Feng’s group has designed a smaller SHANK3 ‘mini-gene’ as a substitute for the full-sized version. Preliminary experiments show that AAV delivery of the mini-gene can rescue phenotypes like anxiety, social behavior and corticostriatal synapse function in SHANK3 knockout mice. Feng also discussed his success in editing the genome in marmosets and macaques using CRISPR/Cas9 technology and showed data from a macaque model of SHANK3 dysfunction25. These models may help test gene therapy approaches and identify biomarkers of brain development closely related to the human disorder.
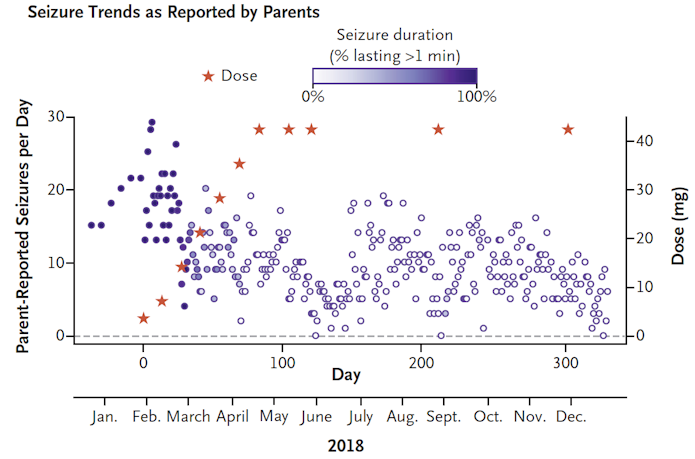
For people with rare conditions brought on by even rarer mutations, individualized gene therapies can provide a pathway for treatment. SFARI Investigator Timothy Yu of Boston Children’s Hospital/Harvard described his ‘N-of-1’ study in treating a girl with Batten disease, a recessive disorder in which a child progressively loses vision, speech and motor control while developing seizures. In a little over a year, an ASO that targeted her unusual splice-site mutation in the CLN7 gene was designed, developed and given intrathecally to the girl26. “The lift was in negotiating with the FDA and working with private organizations, not just in the science,” Yu said. After a year of treatment with the ASO (dubbed ‘milasen’ after the girl, Mila), there were no serious adverse events; seizure frequency and duration had decreased (Figure 5); and possibly her decline had slowed. Though she remains blind, without intelligible speech and unable to walk on her own, she was still attentive and could respond happily to her family’s voices. The highly personalized framework for this drug’s approval is completely different from how medications meant for populations are approved, and it opens a “regulatory can of worms,” Yu said, though he added that the regulators were willing to countenance drug approval for an individual’s clinical benefit.
Rett syndrome
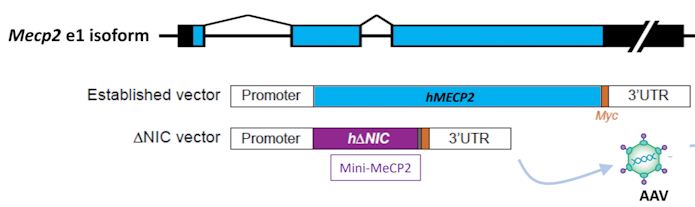
Rett syndrome is a neurodevelopmental condition caused by mutations to the MECP2 gene that has a substantial research base in mouse models. Over 10 years ago, mouse models highlighted the possibility for therapeutics in this condition when Rett-associated phenotypes were rescued by adding back MECP2, even in adulthood27. This reversibility has spurred interest in gene therapy for Rett syndrome, but getting the MECP2 dose right is critical, said Stuart Cobb of the University of Edinburgh and Neurogene: just as too little MECP2 leads to Rett syndrome, too much also results in severe phenotypes. For this reason, it would be nice to package a replacement MECP2 gene with other regulatory elements to control its expression, but this results in constructs that do not fit into viral vectors. To make more room, Cobb and his colleagues have been able to chop away two-thirds of the MECP2, reserving two domains that interact to make a complex on DNA (Figure 6). Mice with this mini-gene are viable and have near normal phenotypes; likewise, injecting this mini-gene into MECP2-deficient mice extended their survival28. Doubling the dose, however, substantially lowered survival. “Putting in safety valves to prevent overexpression is going to be quite important,” he said. One idea is to add back a construct containing only the last two exons of MECP2, which is where most Rett mutations land. These would then be spliced into native transcripts (called ‘trans-splicing’), and thus their expression controlled by endogenous regulatory elements.
Underscoring the double-edged sword of MECP2 dosage, Yingyao Shao from Huda Zoghbi’s lab at Baylor described an MECP2 duplication syndrome (MDS) in humans, which features hypotonia, intellectual disability, epilepsy and autism. Experiments in an MDS mouse model, which carries one mouse version and one human version of MECP2, recapitulates some of the phenotypes of the human condition and can be rescued by an ASO targeting the human allele29. Shao described work to optimize the ASO for translation into humans, which involved developing a more humanized MDS model that carries two human MECP2 alleles. An acute injection of the ASO was able to knock down MECP2 expression in a dose-dependent manner in these mice, and RNA levels dropped a week after injection, with protein levels falling a week later. MECP2 target genes also normalized their expression level, and one maintained this for at least 16 weeks post-injection. The ASO also rescued behavioral phenotypes of motor coordination and fear conditioning, but not of anxiety; these corrections followed the molecular effects, and these timelines would be important to keep in mind while designing clinical trials. Shao also noted that overtreatment with the ASO resulted in Rett-associated phenotypes, but that this was reversible, which suggests that some fine-tuning of dosing in humans might be possible.
To avoid overtreatment and toxicity of any MDS-directed therapy, Mirjana Maletic-Savatic, also at Baylor, is leaving no stone unturned in a hunt for MDS biomarkers that can predict, in each individual, the safety of a particular dose and regimen. Such biomarkers would also help monitor individuals during treatment, give information about target engagement and identify candidates for a particular treatment. Anything found to be sensitive to expression levels of MECP2 could also be useful for Rett, though she noted that MECP2 levels measured in blood do not track linearly with gene copy number. Thus, because of interindividual variability, her approach is to collect a ‘kitchen sink of data’ deriving composite biomarkers that accurately reflect the stage and severity of disease in a given case. She and her colleagues are collecting clinical, genetic, neurocircuitry (such as EEG and sleep waves), immunology and molecular data detected in blood, urine and CSF. These measures are also being explored in induced neurons derived from skin samples of people with MDS. She highlighted two interrelated potential biomarkers in the blood of those with this condition; both measures are downstream targets of MECP2 and are responsive to ASO treatment.
Early detection
Highlights from “Early detection and clinical trial issues” talks and panel discussion, chaired by Paul Wang of SFARI
- There is a need for objective measures to track: clinical endpoints, risk, status, prognosis, safety, pharmacological response or predict response, or diagnose a condition or subgroup within a condition.
- Newborn screening for genetic conditions will help populate early clinical trials of gene therapy.
- To find meaningful clinical endpoints, researchers need to engage with families to understand the areas they would most like to see improvement in for their affected child. Clinical endpoints also have change within the time frame of a clinical trial.
Coming up with objective measures of a person’s status — either their eligibility for a treatment, or whether the treatment has engaged with its target or even whether the treatment is effective — is a real necessity in autism-related conditions, which comprise multiple interrelated behaviors. Eye-tracking methodology may provide such a marker, argued SFARI Investigator Ami Klin of Emory University. Focusing on the core social challenges of autism, Klin, Warren Jones and colleagues have been studying children as they view naturalistic social scenes to quantify their social attention patterns. This has revealed how remarkably early in development social visual learning begins and that this process is disrupted in infants later diagnosed with ASD prior to features associated with the condition appearing. By missing social cues, “autism in many ways creates itself, moment by moment,” Klin said. In considering gene therapy, it may be useful to know that eye looking (how much a subject looks at a person’s eyes, an index of social visual engagement) in particular and social visual engagement in general are under genetic control30; that eye-tracking differences emerge as early as 2–6 months of age; and that homologies in social visual engagement exist between human babies and nonhuman infant primates.
In getting to a point to test gene therapies, identifying those who need them is essential. Wendy Chung of Columbia University and the Simons Foundation illustrated how diagnosis is yoked closely to therapy. To illustrate this, she described her pilot study of newborn blood spots to screen for SMA; at the start, no treatment was available, but the screen identified newborns for a clinical trial of nusinersin. Notably, the screen only cost an additional 11 cents per baby. In the three years since her pilot screen began, the FDA approved two gene therapies for SMA and the SMA screen was adopted for nationwide newborn screening. Currently she is piloting a screen for Duchenne muscular dystrophy and plans to develop a platform that will allow researchers to add other conditions. In prioritizing genetic conditions for gene therapy, she outlined some ideas for focus, such as genes resulting in phenotypes that would not be identified early without screening, those that are relatively frequent, those that are lethal or neurodegenerative, those with a treatment in clinical trials or with FDA-approved medications, and those conditions that are reversible.
In the meantime, Chung also outlined SFARI’s involvement in establishing well-characterized cohorts of individuals with autism, which can help lay a groundwork for gene therapy. People with an ASD diagnosis can join SPARK (Simons Foundation Powering Autism Research for Knowledge), which collects medical, behavioral and genetic information (through analysis of DNA from saliva, at no cost to the participant). If a de novo genetic variant is found in one of ~150 genes, that person is referred to Simons Searchlight, which fosters rare conditions communities and which is also compiling natural history data on people with these mutations.
Clinical trial issues
Bryan King of UCSF discussed how current trial designs for ASD were inadequate for gene therapy trials. As ASD prevalence has grown, parallel design trials — with one group receiving an experimental medicine and the other a placebo — are the standard, but these won’t be possible for the rare conditions that are candidates for gene therapy. Also, change is hard to capture, given the malleable nature of ASD: with no intervention, diagnosis can shift between ASD and pervasive developmental disorder-not otherwise specified (PDD-NOS) in 12–84 months (as defined by the DSM-IV). Current scales are subjective and may miss specific items of clinical significance. (Last year, SFARI funded four efforts to develop more sensitive outcome measures.) King outlined other pitfalls in ASD clinical trials, including significant placebo responses, inadequate sample sizes and not being specific enough when asking about adverse effects. King also mentioned improvements that may arise from just enrolling in a study, which could prompt previously housebound families to venture out with their child, which could kick off a cascade of positive effects. He reiterated how, for gene therapy, a natural history comparison group may be more appropriate, combined with solid outcome measures.
SFARI Investigator James McPartland of Yale University then underlined the need for objective biomarkers for clinical trials, for which there are currently none that are FDA qualified for ASD. As the director of the Autism Biomarkers Consortium for Clinical Trials (ABC-CT), he works with other scientists to develop reliable biomarkers that can be scaled for use in large samples across different sites. McPartland noted a biomarker studied in the ABC-CT: an event-related potential (N170) to human faces, which is on average slower in ASD than in typically developing children. He is working on ways to make it easier for people with ASD and intellectual disabilities to participate in biomarker studies and to make them more socially naturalistic. In discussion, he mentioned he thought it would be possible to look for these kinds of biomarkers in younger children.
SFARI Investigator Shafali Jeste of the University of California, Los Angeles recounted her experience in working with children with genetic syndromes associated with neurodevelopmental conditions. Though she is asked to participate in clinical trials for these conditions, she senses the field has some work to do to be ready for these trials, particularly in those with additional challenges such as epilepsy and intellectual disability. Meaningful and measurable clinical endpoints are still insufficient, and there needs to be more ways to improve accessibility of these trials for these rare conditions. This means developing new measures, such as gait-mat technology that senses walking coordination, or EEG measures in waking and sleep, which have been applied to people with chromosome 15q11.2-13.1 duplication (dup15q) syndrome, who have severe intellectual disability and motor impairments. Jeste also emphasized that increasing remote access to some measures can make a big difference for a trial; for example, a trial of a behavioral intervention for tuberous sclerosis complex that required weekly lab visits was disappointingly under-enrolled until researchers revamped it so most of the intervention could be done remotely31.
Concluding remarks
By grappling with the challenges to gene therapy for ASD, the workshop marked out a faint road map of a way forward. As the scientific questions are answered, the regulatory and clinical trial infrastructure will need to develop apace, and coordination between private, academic and advocacy sectors will be essential. But as gene therapy for diverse human conditions continues to be explored and gene discovery in ASD continues, there is reason to believe that some forms of ASD can eventually benefit from this strategy.
“This workshop provided a terrific discussion about the challenges in developing targeted gene interventions and their potentially transformative effects as therapies,” said John Spiro, Deputy Scientific Director of SFARI. “We are grateful to all the participants, and SFARI looks forward to translating these discussions into focused funding decisions in the near future.”
References
- Mendell J.R. et al. N. Engl. J. Med. 377, 1713-1722 (2017) PubMed
- Satterstrom F.K. et al. Cell 180, 568-584 (2020) PubMed
- Grove J. et al. Nat. Genet. 51, 431-444 (2019) PubMed
- Sanders S.J. et al. Genome Med. 12, 36 (2020) PubMed
- Parikshak N. et al. Nature 540, 423-427 (2016) PubMed
- Jaganathan K. et al. Cell 176, 535-548 (2019) PubMed
- Gilbert L.A. et al. Cell 154, 442-451 (2013) PubMed
- Yen H.S. et al. Science 322, 918-923 (2008) PubMed
- Norman T.M. et al. Science 365, 786-793 (2019) PubMed
- Deverman B.E. et al. Nat. Biotechnol. 34, 204-209 (2016) PubMed
- Chan K.Y. et al. Nat. Neurosci. 20, 1172-1179 (2017) PubMed
- Luoni M. et al. bioRxiv (2019) Preprint
- Huang Q. et al. PLoS One 14, e0225206 (2019) PubMed
- Matharu N. et al. Science 363, eaau0629 (2019) PubMed
- Jüttner J. et al. Nat. Neurosci. 22, 1345-1356 (2019) PubMed
- Schubert R. et al. Nat. Protoc. 14, 3205-3219 (2019) PubMed
- National Academies of Sciences, Engineering, and Medicine. Exploring the scientific opportunities afforded by new nonhuman primate models: Proceedings of a workshop. Washington, DC: The National Academies Press (2019) Report
- Spratt P.W.E. et al. Neuron 103, 673-685 (2019) PubMed
- Huang H.S. et al. Nature 481, 185-189 (2011) PubMed
- Le Fevre A. et al. Am. J. Med. Genet. A 173, 753-757 (2017) PubMed
- Silva-Santos S. et al. J. Clin. Invest. 125, 2069-2076 (2015) PubMed
- Vossaert L. et al. Prenat. Diagn. 38, 1069-1078 (2018) PubMed
- Massaro G. et al. Nat. Med. 24, 1317-1323 (2018) PubMed
- Mei Y. et al. Nature 530, 481-484 (2016) PubMed
- Zhou Y. et al. Nature 570, 326-331 (2019) PubMed
- Kim J. et al. N. Engl. J. Med. 381, 1644-1652 (2019) PubMed
- Guy J. et al. Science 315, 1143-1147 (2007) PubMed
- Tillotson R. et al. Nature 550, 398-401 (2017) PubMed
- Sztainberg Y. et al. Nature 528, 123-126 (2015) PubMed
- Constantino J.N. et al. Nature 547, 340-344 (2017) PubMed
- Hyde C. et al. J. Neurodev. Disord. 12, 3 (2020) PubMed
- From parent advocate to nonprofit chief science officer, to biotherapeutic company cofounder — A personal journey through drug development for Angelman syndrome
- Autism genetics: Where have we been and where are we going?
- New findings in autism research presented at the SFARI spring 2019 science meeting


