Simons Foundation
chevron-down--small
Program Areas
Mathematics & Physical Sciences
Life Sciences
Autism Research Initiative
Neuroscience
Simons Collaborations
Flatiron Institute
Science, Society & Culture
Multidisciplinary Programs
Funding
Funding Opportunities
Policies and Procedures
Award Instructions
Simons Award Manager (SAM) FAQs and Videos
News
Announcements
Articles
Videos
What We’re Reading
Events
Presidential Lectures
Presents
About
About Us
Annual Report
Leadership
Our History
Media Relations
Careers
Summer at Simons
Visitor Information
Contact Us
SFARI
chevron-down--small
SFARI
Funding
Funding Opportunities
SFARI Scientific Perspectives
Fellows-to-Faculty
Conferences and Courses Awards
Grant Policies and Procedures
Simons Award Manager (SAM) FAQs and Videos
Acknowledgment Policy
Sign Up for RFA Announcements
Resources
Autism Cohorts
Autism Inpatient Collection
Simons Searchlight
Simons Simplex Collection
SPARK
Models
iPS cell models
Mouse models
Rat models
Zebrafish models
Other Resources
Autism BrainNet
Research Match
SFARI Base
SFARI Gene
Data analysis tools
Simons Sleep Project
Research
SFARI Investigators
Funded Projects
Autism Rat Models Consortium
Funded Centers
Funded Publications
Research Highlights
News
SFARI News
SFARI Conversations
Workshop and Meeting Reports
Newsletter Archive
Events
All Events
Lectures
Webinars
SFARI Socials
About
About SFARI
Letter from Leadership
Contact Us
Career Opportunities
Our Team
Advisors
SFARI Scientific Advisory Board
SFARI Scientific Review Board
Tag: macrocephaly
All (16)
News (6)
Projects (10)
November 09, 2023
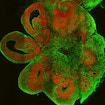
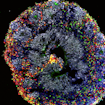
Two studies using brain organoids reveal cell-type-specific changes during the early stages of human forebrain neuronal development in autism
By Azra Jaferi
February 24, 2022
Human derived 3D brain cultures provide insights into brain size changes in 16p11.2 conditions
By Kris Dickson
January 28, 2022
SFARI hosts virtual fall 2021 science meeting
By Michele Solis
September 01, 2016
A novel transcriptional cascade involved in brain overgrowth in autism
September 01, 2016
Convergent signaling pathways linking PTEN and MeCP2, two risk genes for autism spectrum disorders
December 04, 2015
Brain imaging and cell signaling: Insights into the biology of autism
Previous Page
Viewing
Page
1 of 3
Page
2 of 3
Page
3 of 3
Page
1 of 3
Page
2 of 3
Page
3 of 3
Next Page
Subscribe to our newsletter and receive SFARI funding announcements and news
Continue
Funding
Funding
Funding Opportunities
SFARI Scientific Perspectives
Fellows-to-Faculty
Conferences and Courses Awards
Grant Policies and Procedures
Simons Award Manager (SAM) FAQs and Videos
Acknowledgment Policy
Sign Up for RFA Announcements
Resources
Resources
Resources
Autism Cohorts
Autism Cohorts
Autism Inpatient Collection
Simons Searchlight
Simons Simplex Collection
SPARK
Models
Models
iPS cell models
Mouse models
Rat models
Zebrafish models
Other Resources
Other Resources
Autism BrainNet
Research Match
SFARI Base
SFARI Gene
Data analysis tools
Simons Sleep Project
Research
Research
SFARI Investigators
Funded Projects
Autism Rat Models Consortium
Funded Centers
Funded Publications
Research Highlights
News
News
SFARI News
SFARI Conversations
Workshop and Meeting Reports
Newsletter Archive
Events
Events
All Events
Lectures
Webinars
SFARI Socials
About
About
About
About SFARI
Letter from Leadership
Contact Us
Career Opportunities
Our Team
Advisors
Advisors
SFARI Scientific Advisory Board
SFARI Scientific Review Board