
The Simons Foundation Autism Research Initiative (SFARI) held its thirteenth science meeting April 8–12, 2018. Bringing together over 150 SFARI Investigators, collaborators and foundation staff, the gathering provided the occasion to discuss recent results and future directions of SFARI-funded research projects and other studies relevant to autism spectrum disorder (ASD).
Keynote presentations
On the opening night of the meeting, Jonathan Cohen presented the keynote address, which focused on cognitive control, the capacity to guide behavior according to internal goals and overcoming competing intentions. Taking a neurocomputational perspective, he showed that cognitive control entails the trade-off between shared representations that allow learning efficiency and separate representations that enable multitasking and performance efficiency. While stressing the importance of both types of representations in regulating all aspects of human behavior, Cohen speculated that alterations of these learning networks may be implicated in brain disorders such as schizophrenia and ASD. He thus argued for a research approach that not only investigates the individual components of cognition and behavior but also addresses the neural mechanisms that integrate them.
The Monday night keynote presentation was by Matthew Siegel, who drew on findings from the Autism Inpatient Collection1 to discuss some of the challenges of severely affected individuals with ASD. Such challenges include unpredictable aggressive behaviors, nonverbal or low verbal skills and intellectual disability. He showed that wearable devices can help predict the occurrence of aggressive behaviors and provide caregivers with tools to tackle these unpredictable outbursts proactively. His talk brought into the spotlight a subset of the ASD population that is rarely studied and helped to increase awareness about this cohort and the data and biospecimens that are currently available via SFARI Base.
Session presentations
Ten SFARI Investigators presented their latest research findings during the three sessions on Molecular Mechanisms, Circuits, Systems, Cognition & Behavior and Clinical Studies.
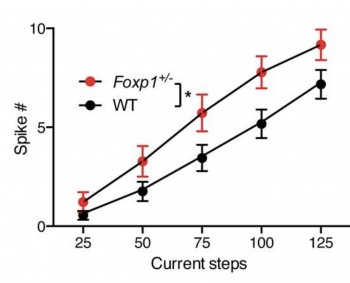
Opening the session on Molecular Mechanisms, Genevieve Konopka presented her laboratory’s work on FOXP1, a transcription factor whose dysregulation has been associated with ASD. She showed that Foxp1 reduction in mice disrupts striatal functions2 and proposed a mechanism whereby the Foxp1 gene defines the molecular identity and regulates the activity of dopamine receptor 2-expressing medium spiny neurons (Figure 1).
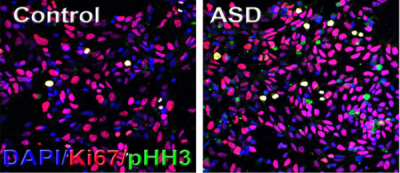
Gaia Novarino then presented her work on SETD5, a high-confidence autism risk gene. Setd5 heterozygous mice exhibited developmental and behavioral abnormalities, accompanied by abnormal histone H4-acetylation and altered gene expression, indicating an important role for epigenetic mechanisms in ASD. The next speaker in this session, Anthony Wynshaw-Boris, discussed the role of the Wnt signaling pathway in early brain overgrowth, a phenotype that has been associated with approximately 20–30 percent of individuals with ASD. He reported that human induced pluripotent stem cell lines derived from ASD individuals with early brain overgrowth display greater proliferation of neural progenitors because of dysregulation of a β-catenin/BRN2 transcriptional cascade3 (Figure 2).
Concluding the session, Beth Stevens shifted the focus from neurons to glial cells; specifically, she discussed the heterogeneity of microglia and autism-relevant perturbations that may affect microglial development and specification.
Moving from molecular mechanisms to system-level dysfunction, the session on Circuits, Systems, Cognition & Behavior discussed using rodent models to study neural and behavioral alterations associated with mutations in high-confidence ASD risk genes. Garret Stuber described distinct neuronal populations in the hypothalamus that respond to social stimuli4 (Video 1) and discussed his ongoing studies looking at neural circuits of social sensory processing in Shank3 knockout mice. Arguing that ASD presents not only with social but also with perceptual impairments, Charles Gilbert then described the circuits integrating primary and secondary visual areas and described his ongoing experiments that assess perceptual performances among several different mouse models of ASD.
With a focus on subcortical circuits, Ann Graybiel presented her recent work delineating the functional contributions of the distinct compartments of the striatum5 — striosomes and matrix — and their developmental origins6. She also discussed potential impairments in striosome-matrix balance in mouse models of ASD. Finally, Peter Kind described an Fmr1 knockout rat model for fragile X syndrome, which has defects in hippocampal synaptic plasticity and impaired episodic memory7. By showing differences and similarities between this rat model and previous Fmr1 mouse models, he argued for the value of studying more rat models in ASD research, so that neural dysfunction, behavior and potential therapeutic treatments can be assessed across species that differ in the complexity of their social behavior repertoires.
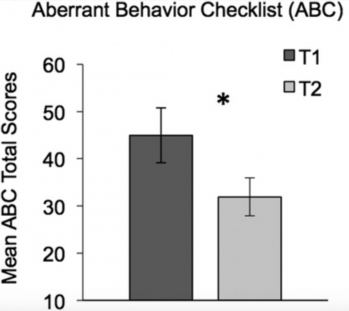
The last session of the meeting focused on Clinical Studies. Rebecca Jones showed that smartphones and wearable devices can provide quantitative measures of behavior, physiology (e.g., arousal) and language aspects of the autism spectrum in both the clinical setting and the home8. Of note, she also reported that, by the end of an eight-week study assessing several outcome measures, caregivers of children with ASD scored their child’s symptoms less severely9 (Figure 3). As no treatment was given as part of the study, these findings demonstrate that placebo-like effects can add considerable confounds and need to be carefully considered when designing clinical trials and evaluating caregivers’ assessments of behaviors. The final presentation in this session focused on another key priority for the field, that is, the development of biomarkers for ASD. Discussing his previous work on a mouse model of social deficits (BTBR mice)10, Elliott Sherr proposed the potential use of blood-based kinase and phosphatase measures during early development.
SCN2A panel
At the previous SFARI science meeting in October 2017, a panel discussing the biology of a single autism risk gene, CHD8, was held. The panel discussed different experimental approaches (e.g., mouse models, induced pluripotent stem cells) to study a genetically defined subgroup of ASD11. Continuing this trend, the April 2018 meeting convened a panel dedicated to another high-confidence autism risk gene, SCN2A, which, together with CHD8, is one of the most recurrent genetic hits in ASD. SCN2A is also of significant research interest for a number of other reasons. These include the fact that this gene has well-defined genotype-phenotype relationships, and that it encodes the α subunit of the Nav1.2 voltage-gated sodium channel, which, like other ion channels, may provide a ‘druggable’ target. Furthermore, over 120 individuals with SCN2A mutations are currently registered as part of the Simons Variation in Individuals Project (Simons VIP)12, thus providing a research-ready cohort for clinical studies.
SCN2A is one of the more promising autism-risk genes...and we have a lot more to learn about the fundamental biology.
- John Spiro, SFARI Deputy Scientific Director
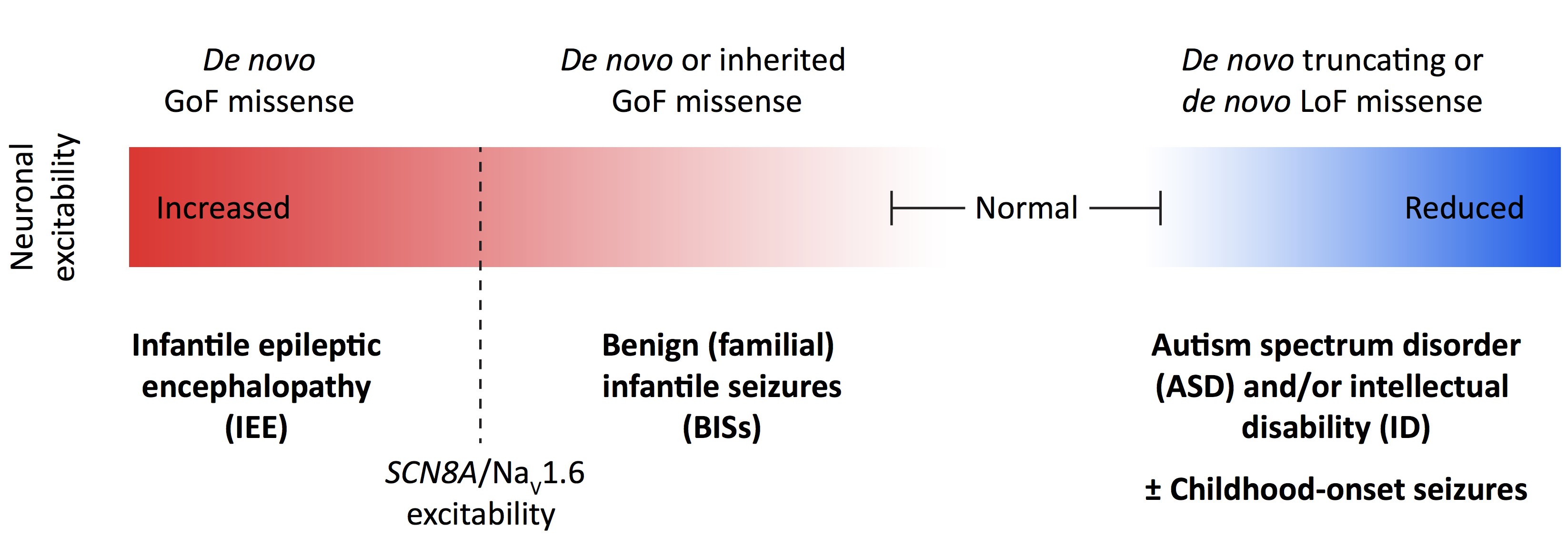
Led by Stephan Sanders, Raphe Bernier, Al George and Kevin Bender, the panel discussed what is currently known about the molecular and cellular biology of SCN2A as well as the phenotypic manifestations observed in individuals carrying mutations in this gene. The speakers also presented recent studies showing that gain-of-function mutations in SCN2A are associated with seizure disorders (specifically, epileptic encephalopathy and benign infantile seizures) while loss-of-function mutations typically associate with ASD13 (Figure 4). Many of these findings are summarized in a recent review article14 coauthored by the speakers and others from academia, industry and SCN2A family advocacy groups, all of whom participated in a workshop organized by SFARI on this topic in April 2017.
“I was extremely pleased that we were able to continue the discussion that was started at the workshop last year and to extend it to this broader audience,” says John Spiro, SFARI Deputy Scientific Director. “SCN2A is one of the more promising autism-risk genes in terms of thinking about potential targeted interventions, and we have a lot more to learn about the fundamental biology.”
Genetics panel
Moving the focus from particular genes to the broader genetic architecture of the disorder, the second panel of the meeting discussed the current state and future directions of ASD genetics research. Matthew State, Joseph Buxbaum, Evan Eichler, William Fairbrother and Olga Troyanskaya reviewed the accomplishments made in the last decade and acknowledged the role played by the Simons Simplex Collection (SSC)15 in shifting the field from hypothesis-driven, single-gene studies to an unbiased gene-discovery approach. The SSC proved to be particularly valuable in identifying the important role that rare de novo mutations play in ASD (Video 2).
The speakers also presented new findings from whole-exome and whole-genome sequencing studies, including variants in noncoding regions of the genome. They agreed that, although several hundred autism risk genes have now been identified, the field has not reached a saturation point; not only is the biology of these genes still mostly unknown, but also our understanding of potential convergent pathways and shared etiology with other neurodevelopmental and psychiatric disorders is still in its infancy.
There was also a consensus that the advent of new technologies and a research approach valuing data-sharing will be key to driving future discoveries. This is especially true for enabling the identification of noncoding regions of the genome that contribute to risk for ASD. While several studies have revealed elevated noncoding regulatory mutation burden in ASD16-19, larger sample sizes will be needed before a complete picture of noncoding variation in ASD emerges20, 21.
“Year after year, I find these meetings ever more inspiring and thought-provoking,” says Louis Reichardt, SFARI Director. “I was very impressed with the quality of the studies presented, and I hope this research will have a long-lasting impact in the autism field and really help accelerate our understanding of the disorder.”
For additional information about SFARI-funded investigators and research projects, please visit: https://www.sfari.org/research/investigators.

Attendees mingle at the meeting’s inaugural reception.


Louis Reichardt, SFARI Director, welcomed attendees during his opening remarks.

James Simons, Chairman of the Simons Foundation, welcomed attendees.

Keynote speaker Jonathan Cohen, Princeton University, talked about cognitive control.

Keynote speaker Matthew Siegel of Maine Medical Center Research Institute described the use of digital, wearable devices in ASD individuals with challenging behavioral issues.

Genevieve Konopka of the University of Texas Southwestern described her research on striatal dysfunction in Foxp1 mutant mice.

Gaia Novarino of the Institute of Science and Technology Austria presented her findings on a role for SETD5 in ASD.

Beth Stevens of Boston Children’s Hospital discussed her laboratory’s latest studies of microglial development.

Investigators share ideas in between sessions.


Ann Graybiel of Massachusetts Institute of Technology presented her latest research findings on the development of striatal compartment architecture and circuitry.

Kevin Bender (University of California, San Francisco), Stephan Sanders (University of California, San Francisco), Raphe Bernier (University of Washington) and Al George (Northwestern University) discussed the biology and behavioral consequences of mutations in SCN2A.

Evan Eichler (University of Washington), Olga Troyanskaya (Simons Foundation, Princeton University), William Fairbrother (Brown University), Joseph Buxbaum (Icahn School of Medicine at Mount Sinai) and Matthew State (University of California, San Francisco) discussed the current state and future challenges of genetics research in ASD.
Attendees mingle at the meeting’s inaugural reception.
References
- Siegel M. et al. Mol. Autism 6, 61 (2015) PubMed
- Araujo D.J. et al. Genes Dev. 29, 2081-2096 (2015) PubMed
- Marchetto C. et al. Mol. Psychiatry 22, 820-835 (2017) PubMed
- Bloem B. et al. Elife 6, pii: e32353 (2017) PubMed
- McHenry A. et al. Nat. Neurosci. 20, 449-458 (2017) PubMed
- Kelly S.M. et al. Neuron 99, 345-361 (2018) PubMed
- Till S.M. et al. Hum. Mol. Genet. 24, 5977-5984 (2015) PubMed
- Jones R.M. et al. NPJ Digit. Med. 1, 34 (2018) Article
- Jones R.M. et al. Autism Res. 10, 1567-1572 (2017) PubMed
- Faridar A. et al. Mol. Autism 5, 57 (2014) PubMed
- Bernier R. et al. Cell 158, 263-276 (2014) PubMed
- Simons VIP Consortium. Neuron 73, 1063-1067 (2012) PubMed
- Ben-Shalom R. et al. Biol. Psychiatry 82, 224-232 (2017) PubMed
- Sanders J. et al. Trends Neurosci. 41, 442-456 (2018) PubMed
- Fischbach G.D. and Lord C. Neuron 68, 192-195 (2010) PubMed
- Turner T. N. et al. Am. J. Hum. Genet. 98, 58-74 (2016) PubMed
- Turner T.N. et al. Cell 171, 710-722 (2017) PubMed
- Zhou J. et al. bioRxiv (2018) Preprint
- Brandler W.M. et al. Science 360, 327-331 (2018) PubMed
- Werling M. et al. Nat. Genet. 50, 727-736 (2018) PubMed
- Wray N.R. and Gratten J. Nat. Genet. 50, 635-637 (2018) PubMed


