Previous studies have shown that disrupting Shank3 function leads to decreased activity in glutamatergic neurons in the hippocampus and cortex, but there were also hints that inhibitory neuron function might be affected. A new paper reported that a nonsense mutation in Shank3 associated with schizophrenia, called R1117X, affects the activity of inhibitory neurons in the cortex, leading to enhanced activity of excitatory neurons and altered behavior in mice.
The work was supported in part by an Explorer Award and an ongoing Pilot Award to SFARI Investigator Alex Kwan. Kwan and his collaborators used two-photon in vivo calcium imaging in medial prefrontal cortex (mPFC) of R1117X mutant mice, isolating calcium transients in apical dendritic spines in layer one that result from subthreshold synaptic input. They found elevated spontaneous calcium transients in dendritic spines and cell bodies of layer two/three pyramidal neurons of R1117X mutants compared to wild-type littermates. Interestingly, they reported the opposite result in a Shank3b deletion mouse thought to more closely model mutations associated with autism spectrum disorder (ASD).
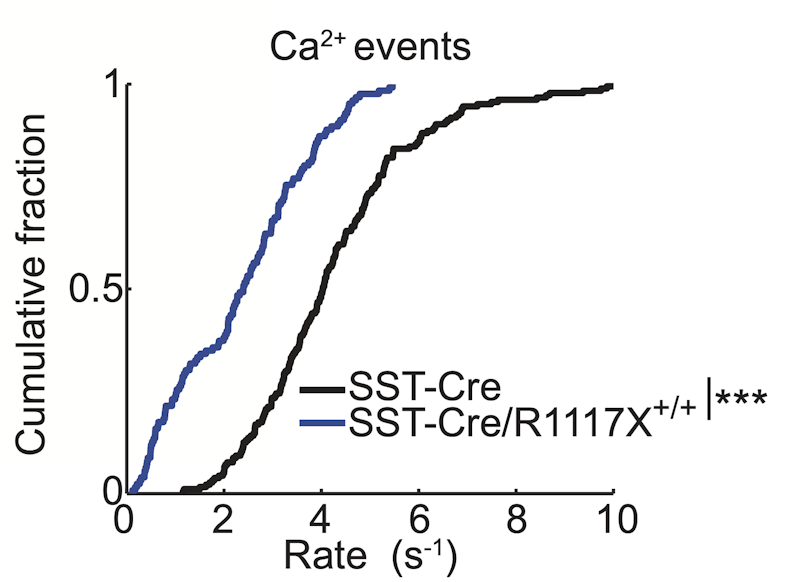
When the investigators looked at interneurons expressing the marker somatostatin (SST+), which make inhibitory synapses on dendritic spines of pyramidal neurons, they found reduced calcium activity in SST+ interneuron cell bodies and axonal boutons in the R1117X mutant mice compared to wild-type littermates. In brain-slice recordings, they also described reduced NMDA receptor-mediated synaptic currents in SST+ interneurons, specifically in the portion of the current mediated by the GluN2B subunit of the NMDA receptor. Viral overexpression of GluN1 and GluN2B in SST+ interneurons increased NMDA currents and calcium activity in SST+ interneurons and decreased calcium transients in dendritic spines of pyramidal neurons in layer one, thus rescuing the mutant phenotype.
Viral overexpression of GluN1/2B in SST+ interneurons in the cortex also rescued the deficit in trace fear conditioning, a behavior requiring mPFC function, seen in R1117X mutants. Changes in prepulse inhibition were also rescued in the mutant mice. However, other behavioral phenotypes, including increased social dominance, anxiety-like behavior or hypo-locomotion, were not rescued, suggesting that these behaviors may be mediated by the function of Shank3 in other brain regions.
The results of this study indicate that various mutations in Shank3 may affect diverse types of neurons differently, potentially with disparate outcomes for the cortical microcircuit and, more broadly, for disease. In future studies, Kwan and colleagues plan to study mice harboring another mutation in Shank3, InsG3680, which has been associated with ASD, as well as mice with loss-of-function in Scn2a, another high-confidence ASD risk gene
Reference(s)
Inhibitory regulation of calcium transients in prefrontal dendritic spines is compromised by a nonsense Shank3 mutation.
Ali F., Shao L.X., Gerhard D.M., Sweasy K., Pothula S., Pittenger C., Duman R.S., Kwan A. C.


