Changes in genes that regulate mRNA translation in neurons have been identified as risk factors for autism spectrum disorder (ASD) (Louros and Osterweil, J. Neurochem., 2016). In one well-studied example of this, mutations in the FMR1 gene give rise to the most commonly identified single-gene cause of ASD — fragile X syndrome (FXS). Mutations in the FMR1 gene result in a loss of the RNA-binding protein FMRP, which has a well-described role in normally suppressing the translation of mRNA into protein (Laggerbauer et al., Hum. Mol. Genet., 2001). Many of FMRP’s targets encode critical synaptic proteins (Darnell et al., Cell, 2011), making it a key player in brain development and synaptic plasticity.
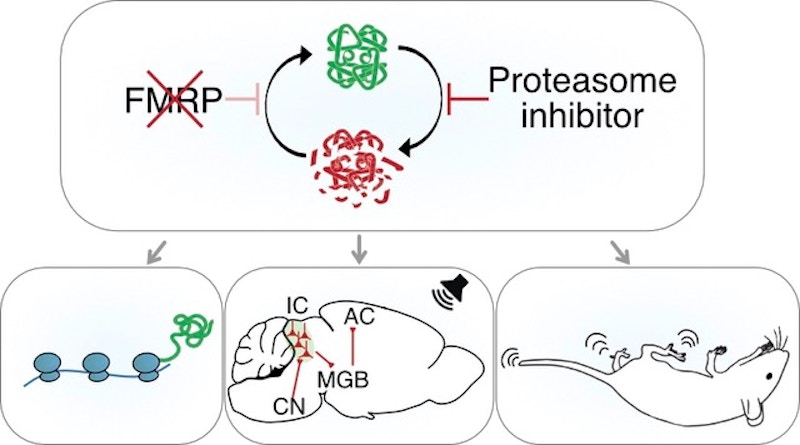
In line with FMRP’s role as a translational suppressor, excessive neuronal protein synthesis has been reported in animal models missing the FMR1 gene as well as in cells from individuals with FXS (Richter et al., Nat. Rev. Neurosci., 2015). These findings suggested that a harmful build-up of proteins is a major contributor to the phenotypes of FXS. However, there have been observations of a mismatch in protein synthesis and protein accumulation — an overall increase in protein synthesis has been observed, but not a general accumulation of proteins at synapses (Seo et al., Nat. Commun., 2022). One reason for the lack of protein accumulation could be a compensatory increase in protein degradation, as hypothesized by Emily Osterweil. In a recent study supported in part by the Simons Initiative for the Developing Brain (SIDB), Osterweil and her colleagues showed that the increased protein synthesis in FXS drives a pathological compensatory rise in protein degradation, which can be targeted to correct various deleterious changes in FXS (Louros et al., Neuron, 2022).
Osterweil and her colleagues looked at protein synthesis and degradation in hippocampal neurons of a mouse model of FXS (Fmr1−/y) and wildtype mice. In neurons of the Fmr1−/y mice, they found increases in proteasome activity along with upregulation of the ubiquitin proteasome system (UPS), the major pathway in charge of targeted protein degradation. Consequently, there was an accelerated degradation of proteins in Fmr1−/y hippocampal synapses. The elevated proteasome activity observed in Fmr1−/y neurons was found to be downstream of elevated protein synthesis, suggesting that the two processes are dynamically coupled. They next showed that, in Fmr1−/y mice, normalizing UPS activity (with a proteasome inhibitor, bortezomib) corrected excessive protein synthesis in the hippocampus. It also corrected hyperexcitability in the Fmr1−/y inferior colliculus, which prevented the hyperactivation of these neurons in response to sound. Further, reduction of UPS (either pharmacologically or genetically) prevented audiogenic seizures in Fmr1−/y mice.
In sum, Osterweil and her colleagues show that excessive activity of the UPS pathway contributes to pathological changes in the Fmr1−/y brain and that UPS reduction could be a new approach to correcting multiple phenotypes in FXS, for which novel therapeutic strategies are urgently needed.
Reference(s)
Excessive proteostasis contributes to pathology in fragile X syndrome.
Louros S., Seo S., Maio B., Martinez-Gonzalez C., Gonzalez-Lozano M., Muscas M., Verity N., Wills J., Wan Li K., Nolan M., Osterweil E.


