The transcription factor Tbr1 has been convincingly identified as an autism risk factor, and researchers have previously shown that its function is critical for the typical development of deep-layer cortical neurons. A new study in mice sheds further light on its role in synapse and dendrite development and identifies a number of downstream targets and possible avenues for therapy.
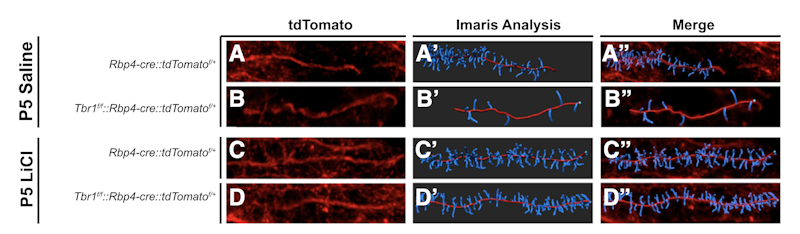
Supported in part by a Research Award to SFARI Investigator John Rubenstein, the work used mouse lines that are constitutively heterozygous for Tbr1, or lack one copy of the gene only in layer 5 or layer 6 neurons of the cortex. Single-cell RNA sequencing of layer 5 Tbr1-deficient neurons identified a number of transcripts whose expression was significantly altered, including Kif1a, Gsk3b, Wnt7b, Ctnnb1, Ank2 and Smarcc2. Additional analyses showed a reduced number of excitatory and inhibitory synapses in deep-layer neurons, reduced dendritic spine density, increased hyperpolarization-activated cation currents and alterations in social behavior. Given previous evidence showing that WNT signaling is Tbr1-dependent and the identification of WNT-related transcripts as Tbr1 targets, Rubenstein and colleagues then asked whether restoring normal WNT signaling could rescue any of these phenotypes. Strikingly, administration of LiCl or a GSK3b inhibitor, both expected to normalize WNT signaling, had substantial effects on synapse and dendritic spine development in deep-layer pyramidal neurons, as well as on social behavior.
The authors proposed a model for Tbr1 function in deep-layer neurons that includes its regulation of WNT signaling, retinoic acid metabolism and kinesin-dependent transport. They concluded by suggesting that LiCl should be rigorously evaluated as a possible therapy for Tbr1-associated syndrome as well as for conditions with overlapping synaptic abnormalities.
Reference(s)
Enhancing WNT signaling restores cortical neuronal spine maturation and synaptogenesis in Tbr1 mutants.
Fazel Darbandi S., Robinson Schwartz S.E., Pai E.L., Everitt A., Turner M.L., Cheyette B., Willsey A. J., State M., Sohal V., Rubenstein J.


