
With the coronavirus pandemic bearing down on the United States, the Simons Foundation Autism Research Initiative (SFARI) decided to host a virtual version of its spring 2020 science meeting. What had been scheduled for three days in April (April 5–7) was instead rolled out in a series of seven weekly webinars held between April 17 and May 29. The webinars included a panel on the role of maternal and immune contributions to autism risk, as well as presentations on the topics of autism genetics, molecular mechanisms and clinical aspects. In all, over 400 SFARI Investigators, collaborators and staff attended.
Panel: Maternal and immune contributions to autism
Though genetic risk factors for autism spectrum disorder (ASD) have been steadily identified, evidence for other factors, such as premature birth, placental insufficiency, infections during pregnancy and signs of immune system activation in postmortem brain tissue1,2 have suggested there are other contributors afoot. Immune responses could somehow interfere with fetal development, but questions remain about whether signs of immune system dysregulation are causes or consequences of ASD, or whether understanding their mechanisms could benefit diagnosis or treatment.
John Lukens of the University of Virginia examined the links between maternal inflammation and behavioral anomalies in offspring in a mouse model of maternal immune activation (MIA). After injecting polyinosinic:polycytidylic acid (polyI:C), a viral mimic, into pregnant mice, Lukens found behavioral changes in the offspring that were sex dependent, a finding that he further explored at the level of transcriptomes of male and female mouse placenta (each fetus in a mouse litter has its own placenta). Notably, over a third of genes that showed sex differences in expression in MIA offspring are denoted as high confidence or strong candidate autism risk genes by SFARI Gene. The pathways affected involved immune function and, at later stages, placental health, which raises the possibility that placenta evaluation during pregnancy may eventually index autism risk.
The placenta can exert its own influence on fetal development through hormones. Anna Penn of Columbia University illustrated this with her work on a placental hormone called allopregnanolone (ALLO), which increases during gestation and is linked to neuronal proliferation and myelination in the brain. She has developed a transgenic mouse model in which different placentas within a litter can have varying levels of ALLO3. At postnatal day 30 (adolescence for mice), she found that male mice that had gestated with an ALLO-insufficient placenta had differentially expressed genes in the cerebellum compared to those that were ALLO sufficient. White matter development was particularly impaired, with an increase in myelin thickness for males but a decrease for females. ALLO-insufficient mice also exhibited behavioral anomalies, such as decreases in sociability, altered gait and increases in repetitive behaviors, but in males only. The work suggests that assessing the state of the placenta during pregnancy, and rectifying any hormonal anomalies therein, might eventually offer a way to mitigate risk to fetal brain development.
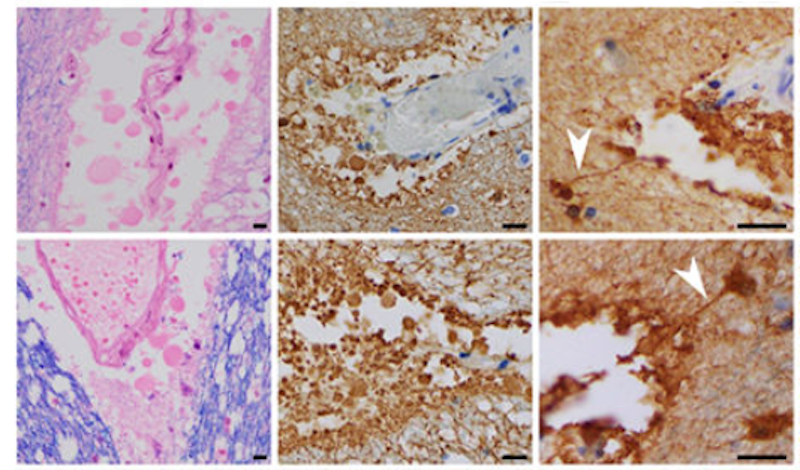
Matthew Anderson of Harvard Medical School and director of Autism BrainNet Boston node presented his recent findings showing new evidence of immune system activation in postmortem brains from people with ASD4. Examining brains donated to Autism BrainNet, he found that T-lymphocytes surrounding vessels to form ‘perivascular lymphocytic cuffs’ were elevated in ASD compared to neurotypical brains (Figure 1). He also described round membranous ‘blebs’ coming off of astrocytes in the autism brains; specifically, these blebs came off of astrocytic foot-like processes that line the vascular space to form the glia limitans. Anderson proposed a neuropathological process whereby cytotoxic T-cells gain entry to the perivascular space surrounding the brain, then attack the glia limitans, which may compromise the blood-brain barrier. He argued that the glial fibrillary astrocyte protein (GFAP) and other proteins entering the cerebrospinal fluid, and ultimately the blood, may provide a first potential biomarker associated with a specific immune pathology in the autism brain.
Clinical
The next webinar focused on clinical issues in autism. Catherine Lord of the University of California, Los Angeles described her evaluation, which was designed to pick up changes in social communication over the time scales used by many clinical trials. Called the Brief Observation of Social Communication Change (BOSCC), the assessment examines interactions during play sessions; it can detect changes over 8 to 12 weeks, does not rely on clinician or parent report and does not require a lot of time to conduct, nor to train a person to conduct. Though the BOSCC is reliable5 and has been used in a growing number of studies, Lord addressed some challenges that have arisen, such as the need to adapt it for older, verbal individuals. In addition, she is working to automate coding of these interactions.
Therapeutic approaches that seem promising in animal models often fail in humans, and this problem is blamed for much of the stalled drug development for central nervous system disorders. To try to improve upon animal models for ASD, Karen Parker of Stanford University studies naturally occurring variations in social skills in a large outdoor colony of rhesus macaques. Rhesus macaques are a very social species, which is also evolutionarily closer to humans than rodents. Ratings of these monkeys using an adapted version of the Social Responsiveness Scale (SRS), which was first developed as an autism screening tool in humans, have identified ‘low-social’ individuals in the general rhesus monkey population.
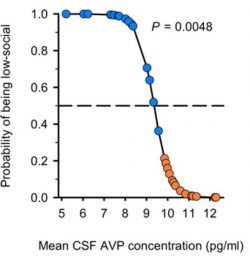
Parker reported that low-social monkeys have lower levels of arginine vasopressin (AVP) in the cerebrospinal fluid (CSF) than high-social monkeys6 (Figure 2). This finding mirrors her studies of AVP in humans, showing lower AVP levels in children with autism compared to children without autism6,7. Combined with recent evidence that AVP levels in the CSF are lower in infants before autism symptoms emerge8, these studies support the possibility that AVP may be a biomarker of sociality in autism. Exploring therapeutic options, Parker also demonstrated that intranasal treatment with AVP improved social interactions in children with autism9; she is now trying to replicate these results in a larger study.
The last speaker of this session, Michael Platt of the University of Pennsylvania, also studies social interactions in rhesus macaques to better understand the relationship between sociality and brain functions. He discussed a population of wild rhesus macaques living on the island of Cayo Santiago. Platt and others have been studying these monkeys for 13 years and have comprehensive data on their life histories, social networks, behavior and cognition, as well as bio-banked tissues, including 368 brains. He described relationships between the neuroanatomy of brain regions that process social information (e.g., superior temporal sulcus and anterior cingulate gyrus) and social behaviors (e.g., the number of grooming partners a monkey had). His previous work has linked social attention to genes related to serotonin production10, and full genome sequencing is now underway for about 250 monkeys to find other genetic contributors to social function. Some of the 19 million variants discovered so far are found in many genes listed in SFARI Gene and are linked to neuromodulation involving serotonin, dopamine, oxytocin and vasopressin.
Molecular mechanisms
Two webinars then discussed the molecular mechanisms that could explain how ASD-related genetic changes affect neural functions. In the first of the ‘molecular’ webinars, Mustafa Sahin of Boston Children’s Hospital and James Ellis of the Hospital for Sick Children discussed their research using human neurons derived from skin cells of individuals with genetic changes through induced pluripotent stem cells (iPSCs).
Sahin studies tuberous sclerosis complex, a rare genetic condition caused by mutations in either the TSC1 or TSC2 genes. This disorder is characterized by noncancerous tumors that grow throughout the body and is often associated with epilepsy and ASD. Sahin argued that these mutations may affect biological pathways that are disrupted in other ASD-related disorders. In Purkinje neurons derived from individuals with tuberous sclerosis complex due to a mutation in TSC2, he found decreases in transcripts of genes that are known targets of FMRP, an RNA binding protein linked to fragile X syndrome, as well as a decrease in FMRP itself11; these differences were particularly pronounced for neurons that had biallelic mutations in TSC2. When iPSCs derived individuals with tuberous sclerosis complex were induced to become excitatory cortical neurons, there was similar downregulation of FMRP target transcripts, but FMRP levels were unchanged12. These findings suggest a complex interplay between TSC2 and FMRP on both transcriptional and translational levels, which may affect neuronal function and lead to the development of ASD.
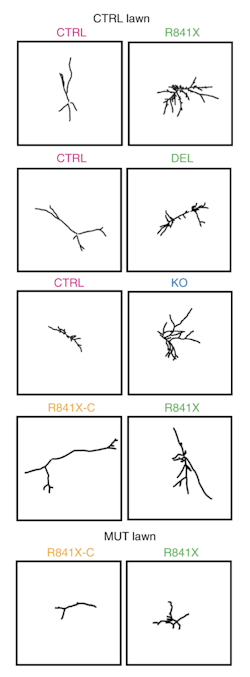
Ellis followed with data on cellular phenotypes of neurons derived from iPSCs made from people carrying mutations in SHANK2, a high-confidence autism risk gene. To examine the morphology and function of these neurons, Ellis used a co-culture system in which neurons carrying mutations of interest are labeled and sparsely grown on a lawn of wild-type neurons13. This revealed excessively long dendrites in SHANK2 mutated neurons — longer than wild-type neurons as well as isogenic control neurons that are genetically identical to the SHANK2 mutant neurons, except that their mutation in SHANK2 had been corrected with gene editing (Figure 3).
Ellis’s lab has also begun to query the state of RNA translation in these types of neural models. In neurons derived from people with Rett syndrome, a condition caused by MECP2 mutations and associated with autism features, he reported fewer transcripts directly engaged with ribosomes compared to wild-type neurons, which indicates a lower level of translation14. Many genes showed this reduction, including NEDD4L, an enzyme that marks proteins with ubiquitin, thus flagging them for degradation. Consistent with compromised degradation, targets of NEDD4L — including potassium and glutamate channels — accumulated in Rett cells. Ellis emphasized that a shift in abundance of these channels was not predicted by their transcripts, which highlights the importance of directly exploring the ‘translatome.’
The second webinar on the topic of molecular mechanisms continued the focus on understanding autism through cellular analyses. James Noonan of Yale University detailed his lab’s work to understand the transcriptional consequences of loss of CHD8, an ASD-risk gene that regulates the expression of other genes. Using single-cell RNA-sequencing to profile the transcriptomes of over 140,000 cells from embryonic mouse brain, his group, in collaboration with Rebecca Muhle, found a smooth progression of expression patterns that recapitulates the stages of cortical neurogenesis: some cells had patterns found in radial glia, others resembled progenitor cells, others had a neuron-like pattern, and still others had patterns in between these cell types. To understand how different genes might work together, Noonan’s lab grouped genes into a number of ‘meta-genes’ which shared similar expression profiles over the course of cortical neurogenesis. CHD8 and other ASD-risk genes belonged to a meta-gene marked by increased expression during the later neural differentiation stage, which suggests this is a particularly sensitive time of fetal brain development.
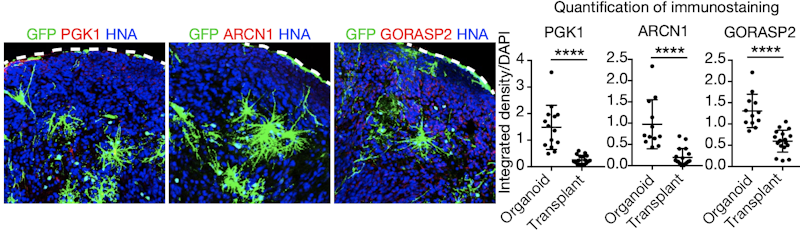
Brain organoids made from human cells have shown much promise for modeling human brain development and the perturbations thought to underlie neurodevelopmental conditions such as ASD. Yet Arnold Kriegstein of the University of California, San Francisco sounded a cautionary note about brain organoids in a talk that compared how well they compare to primary tissue. The good news is that, across numerous different protocols, brain organoids largely reproduce the same broad cell classes found in human fetal brain tissue15. But some key differences emerge: organoid cells do not form layers characteristic of the cortex; they do not accurately reproduce the gene-expression profiles seen in primary tissue; they do not exhibit cell subtype diversity; and their cell identity can appear degraded, gene-expression-wise. Organoid cells also seemed under stress because they upregulated genes in metabolic stress pathways. This could be remedied by transplanting organoid cells into mouse cortex, where stress-related gene expression went down and identity of the cells became ‘crisper’ in terms of gene expression (Figure 4). Kriegstein suspects that the stress may come from cell-culture media, which could be studied and reformulated; in the meantime, understanding organoid shortcomings is important for interpreting their results.
Genetics
Two webinars discussed outstanding questions in ASD genetics. During the first webinar, three talks investigated potential genetic contributors to autism that are outside the conventional genomic search space. Maria Chahrour of the University of Texas Southwestern Medical Center explored whether mobile elements in the genome called retrotransposons might have a hand in autism risk. Mobile genetic elements that can move from place to place in the genome make up a large proportion of the genome’s sequence, but their movement is largely contained. If these containment mechanisms fail, however, a mobile element could move and reinsert itself into the genome, possibly in a place that does some damage — like a gene. Chahrour focused on a class of retrotransposons called long interspersed elements (LINE1). Using publicly available RNA-sequencing data1, her preliminary findings revealed changes in levels of LINE1 transcripts in the visual cortex of individuals with autism relative to individuals without autism, which suggests some LINE1 elements are active. LINE1 encodes two open reading frame proteins that are needed for mobilization; immunohistochemistry detected one of these in the autism postmortem brain at increased levels relative to postmortem brains from donors without autism in the same cortical region. Future work will determine whether changes in LINE1 transcripts are specific to autism, whether an engineered increase in their activity can cause autism-associated phenotypes in mice and whether they might be linked to an inflammatory response.
Jessica Tollkuhn of Cold Spring Harbor Laboratory described her work that aims to understand the roots of the male bias for ASD diagnosis in mouse models. Sex hormone surges happen in the brain prenatally and at birth, in addition to the well-known surge at puberty. Testosterone and estrogen bind to steroid receptors that then move into the nucleus to bind to the genome and act as transcription factors. Any sex-based differences in gene regulation could elevate risk for males or protect females. In postnatal mice, Tollkuhn used single-cell RNA-sequencing of estrogen receptor-expressing neurons, which revealed sex differences in gene expression in highly sexually dimorphic regions of the limbic system. Specifically, genes with higher levels of expression in females than in males included more ASD risk genes than expected; this may somehow afford females some protection to disabling mutation to one copy of these genes. To address how differential gene expression may occur in the first place, Tollkuhn has identified the location of estrogen receptor binding sites in the genome and found ASD risk genes that are among its targets. Next steps include nailing down the locations of androgen and estrogen receptors in the developing mouse brain.
Another potential contributor to autism risk may reside within mitochondrial DNA (mtDNA). Given that autism has been linked to dysfunctional metabolic processes and given that the brain consumes an outsized amount of the body’s energy, Neal Sondheimer of the Hospital for Sick Children has been looking for genetic variation within mtDNA that may associate with autism. What the mtDNA sequence lacks in length (it contains 37 genes), it makes up for in variety through heteroplasmy, in which different mitochondria within a cell can carry different genetic variants.
Using whole genome sequences from the large MSSNG cohort of over 11,000 people, nearly half with autism, Sondheimer’s survey of mtDNA identified some pathogenic variants causing bona fide mitochondrial disorders, thus affirming a relationship between autism and mitochondrial disorders. His research also identified a rare heteroplasmic risk allele for ASD that occurred more commonly in the MSSNG cohort than in the general population, which suggests that heteroplasmic variants may be a source of genetic risk. Future work will include functional analysis of these mitochondrial variants in cell models.
The next and final webinar in the series featured two speakers intent on finding ways to understand the function of ASD gene variants. Kurt Haas of the University of British Columbia described a collaborative project with the goal of creating a suite of assays that can test the function of many variants in a single gene to build a more refined picture of their impact. This offers a more empirical method than the computational algorithms used now to determine whether a newfound genetic variant is likely to be deleterious to a protein’s function. Haas and his colleagues devised 18 assays in five model systems to test variants to PTEN, which encodes a protein and lipid phosphatase that has been associated with ASD16.
A screen in yeast of known biochemical variants of PTEN developed some useful benchmarks of PTEN function, such as enzymatically inactive or constitutively active proteins. When nearly 50 ASD-associated PTEN variants were observed in yeast alongside these biochemical variants, the assay revealed a wide range of PTEN activity, from loss of function to partial loss of function, to no effect, to gain of function (Figure 5). This variability was confirmed in other assays involving Drosophila growth, rat neuron cultures, chemotaxis by C. elegans and activity of the pAKT signaling pathway in human cells. Notably some ASD-related PTEN variants destabilized the PTEN protein, which correlated with their loss-of-function phenotypes. Integrating this information across assays deemed 38 out of 49 ASD-related PTEN variants to be pathogenic or likely pathogenic, albeit through different molecular mechanisms. Haas said the method could detect partial loss-of-function phenotypes better than computational algorithms did, which tended to rate these as either severe or not affected.

Joseph Dougherty of Washington University described his lab’s pipeline for characterizing genetic variants in the untranslated regions (UTR) surrounding coding sequences in mRNA transcripts. Variants in these areas may alter translation through influencing a transcript’s stability or its ability to be loaded onto a ribosome for translation17. Deficiencies in either could substantially decrease generation of a protein product and potentially result in a loss-of-function phenotype, without a corresponding loss-of-function mutation to the coding sequence. Dougherty has devised ways to characterize thousands of UTR variants at a time, based on unique RNA barcode sequences to keep track of each variant. Preliminary results in cell lines show that most UTR variants — both from cases with and without ASD — have an effect on transcript stability or ribosomal occupancy; other evidence suggested that, at least in vitro, transcript stability may be a driver of ribosome occupancy. In vivo, Dougherty reported that a portion of those mutations, including some from ASD cases, resulted in changes of a gene’s product.
Keynote speaker
On May 29, SFARI welcomed Allyson Berent as the keynote speaker to conclude the SFARI spring 2020 science meeting with a special webinar open to the public. Berent has a young daughter with Angelman syndrome, and as a veterinarian with experience in getting medical devices into clinical trials, she found herself in a position to help expedite the development of therapeutics for Angelman syndrome through translational research. In her talk, she shared the story of her daughter’s diagnosis at five months of age, and her observations of the field as she engaged with researchers, the Foundation for Angelman Syndrome Therapeutics (FAST) and eventually FAST’s own biotech company, GeneTx Biotherapeutics, of which she is chief operating officer.
Angelman syndrome is caused by damage to the maternally inherited copy of UBE3A. Though the body can make all the UBE3A protein product it needs from the paternally inherited copy, the brain cannot — it reads only the maternal copy while the paternal copy remains silent. This leaves the brain to develop without a working copy of UBE3A in Angelman syndrome, which results in lack of speech, seizures, poor motor skills. Berent’s hopes for a therapeutic were galvanized by a study of Angelman mosaicism18, which suggested that even correcting a small percentage of cells might have a strong impact.
As chief scientific officer for FAST, Berent helped prepare for clinical trials by contributing to the launch of the Angelman Biomarker and Outcome Measure Consortium. The group surveyed families to identify the domains most impacted in Angelman syndrome and which caretakers found most meaningful. Based on this, the group developed endpoints better suited for Angelman syndrome. After FAST received the largest single donation in its history to fund various preclinical therapeutic programs, one program based on research by Scott Dindot at Texas A&M University stood out. This program involves an antisense oligonucleotide (ASO) that could un-silence the paternally inherited UBE3A allele in the brain. After securing the license for this technology, FAST launched GeneTx Biotherapeutics to test human ASO candidates. “We felt we had a better chance to stay singularly focused on Angelman syndrome through the early drug development process if we took this on ourselves with key support of world-renowned experts,” Berent said.
GeneTx is now partnering with Ultragenyx for a phase1/2 clinical trial of an ASO in children with Angelman syndrome. In contemplating the miraculous speed in getting this ASO to trial, Berent cautioned against getting hung up on perfecting mouse models: their role was valuable to establish a proof of concept for a therapeutic and to strongly inform dosing and toxicity, but that hyperfocus on specific behaviors or their ability to be rescued could soak up time without translatable benefits.
Concluding remarks
While many missed the spontaneous interactions of the in-person meeting, the virtual format made it possible for the SFARI community to continue its discussion of priorities and new findings in autism research. An added bonus was that members of SFARI labs who would not normally attend the meeting, such as students and postdocs, also had the opportunity to join the webinars.
SFARI director Louis Reichardt concluded the series with some perspectives. “I’m happy the sessions went extremely well, and I hope that all who participated also enjoyed them,” he said. “We look forward to seeing you all again in person soon.”
References
- Gupta S. et al. Nat. Commun. 5, 5748 (2014) PubMed
- Parikshak N.N. et al. Nature 180, 423-427 (2016) PubMed
- Vacher C.-M. et al. bioRxiv (2019) Preprint
- DiStasio M.M. et al. Ann. Neurol. 86, 885-898 (2019) PubMed
- Grzadzinski R. et al. J. Autism Dev. Disord. 46, 2464-2479 (2016) PubMed
- Parker K.J. et al. Sci. Transl. Med. 10, eaam9100 (2018) PubMed
- Oztan O. et al. Ann. Neurol. 84, 611-615 (2018) PubMed
- Oztan O. et al. Proc. Natl. Acad. Sci. USA 117, 10609-10613 (2020) PubMed
- Parker K.J. et al. Sci. Transl. Med. 11, eaau7356 (2019) PubMed
- Watson K.K. et al. Anim. Behav. 103, 267-275 (2015) PubMed
- Sundberg M. et al. Mol. Psychiatry 23, 2167-2183 (2018) PubMed
- Winden K.D. et al. J. Neurosci. 39, 9294-9305 (2019) PubMed
- Zaslavsky K. et al. Nat. Neurosci. 22, 556-564 (2019) PubMed
- Rodrigues D.C. et al. Cell Rep. 30, 4179-4196 (2020) PubMed
- Bhaduri A. et al. Nature 578, 142-148 (2020) PubMed
- Post K.L. et al. Nat. Commun. 11, 2073 (2020) PubMed
- Sapkota D. et al. Cell Rep. 26, 594-607 (2019) PubMed
- Nazlican H. et al. Hum. Mol. Genet. 13, 2547-2555 (2004) PubMed
- A Conversation with SFARI Investigator Karen Parker
- T-lymphocytes target astrocytes in the postmortem autism brain
- From parent advocate to nonprofit chief science officer, to biotherapeutic company cofounder — A personal journey through drug development for Angelman syndrome
- New findings in autism research presented at the SFARI spring 2019 science meeting


