
Karen Parker is an associate professor at Stanford University. Her laboratory studies the biological basis of sociality, with an emphasis on the role of vasopressin and oxytocin signaling pathways.
She has developed a nonhuman primate model to study naturally occurring individual differences in social behavior in rhesus monkeys. Rhesus monkeys are evolutionarily closer to humans than other commonly used model organisms (e.g., rodents) and exhibit a wide range of complex social behaviors. She studies how the neuropeptides arginine vasopressin (hereafter, vasopressin) and oxytocin regulate differences in social behaviors in this species as a point of entry to identifying biomarkers for neurodevelopmental disorders affecting social functioning, such as autism spectrum disorder (ASD). In addition, she conducts translational and clinical research with the goal of developing pharmacological biomarker-informed treatments that can improve social outcomes in individuals with ASD.
To date, she has received four awards from SFARI, including a Pilot Award in 2008 to study oxytocin biology in ASD; a second Pilot Award in 2013 to identify biomarkers of sociality in her non-human primate model; a Research Award in 2015, through which to better characterize the social impairments exhibited by the ‘low-social’ monkeys and to test therapeutic strategies; and a Director Award in 2019 to begin studying infant monkeys at risk for poor social outcomes.
Your laboratory investigates the biology of sociality. Could you talk about your research and how it relates to autism spectrum disorder?
I began studying the biology of social functioning in voles as a graduate student at the University of Michigan. As a postdoctoral fellow in the psychiatry department at Stanford University, I began working with nonhuman primate models, which can provide critical insights that stand to advance our understanding of neurodevelopmental and psychiatric disorders characterized by social impairments.
A lot of times, there’s this pivotal moment in the scientific zeitgeist, if you will, where your career takes a certain turn; for me, that was when I was setting up my new lab as an assistant professor at Stanford. At that critical juncture, Stanford had just formed an autism working group, and I was pulled into discussions with a group of autism clinicians who were interested in speaking with basic scientists to learn more about the biology of social functioning. I formed a collaboration with several of them, and we submitted a proposal for one of the Simons Foundation Autism Research Initiative (SFARI)’s requests for applications (RFAs). That’s really how I became an autism researcher. We started doing work on biomarkers as well as research on therapeutics in people with autism. I then began developing a monkey model because I felt there was a tremendous opportunity to create an animal model that had high translational utility. SFARI was critical in funding this work.
Tells us more about your work with nonhuman primates, and its implications for advancing autism research.
When thinking about points of entry into modeling a disorder, we were interested in establishing face validity (i.e., do the monkeys show behavioral features associated with autism) and construct validity (i.e., do the same biological mechanisms regulate these behaviors in both species, monkeys and humans). There’s very good evidence now that suggests autism and its social features are heritable and that the majority of genetic burden in autism is polygenic. Epidemiological and twin studies also suggest that this polygenicity likely underlies individual differences in social functioning. So we were interested in seeing if we could — as had been done in human populations — target the social behavioral extreme of a large population of monkeys. We started studying monkeys that had naturally occurring low social abilities [compared to those that were socially competent] as a window into investigating the biology of sociality. We then planned to translate these findings from low-social monkeys directly to people with autism.
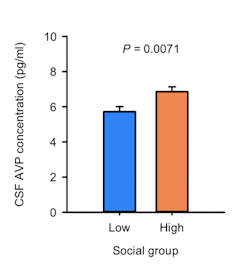
This research led to discoveries of potential biomarkers.
I was interested in identifying biomarkers in monkeys that would have high translational potential. We looked at potential biomarkers in both blood and cerebral spinal fluid (CSF). Significant progress had been made in identifying biomarkers for dementia and multiple sclerosis using a CSF-based approach; I wanted to see if we could get any traction using a similar method for autism.
In the work that I had done in voles as a Ph.D. student, I had been studying vasopressin as a prosocial molecule in males. That was quite interesting to me in retrospect because autism has a sex bias, being more prevalent in boys. We thus initially set out to study vasopressin in male monkeys to see if vasopressin was low in monkeys with social impairments. That was one of the first studies we did1.
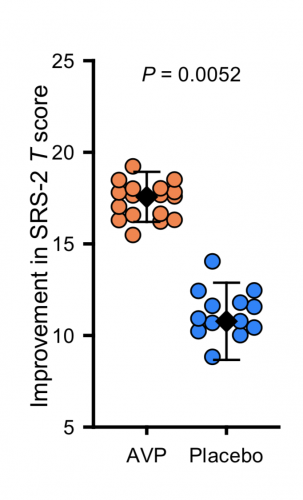
Last year, you published findings from a randomized placebo-controlled pilot trial on vasopressin2, showing improvement of social symptoms following administration of a vasopressin agonist in children with autism. How did you go from a monkey model to humans?
One of the rationales for administering vasopressin was that both in our monkey model and then in two subsequent independent human cohorts, we saw that vasopressin was low in socially impaired monkeys1 and in people with autism3. We also found that the lower the vasopressin levels were in CSF, the greater the social impairments in both monkeys and people1,3. We reasoned that administering vasopressin could increase social abilities in kids with autism. And so, Antonio Hardan and I set out to do a first-in-class pilot treatment trial using a gold standard double-blind randomized placebo-controlled design. Our primary outcome measure was the Social Responsiveness Scale (SRS), 2nd edition, which is a parent report measure. It was also important for us to be able to show that we saw convergent validity between what the parents reported in a blinded fashion, what the clinicians reported in a blinded fashion and what the kids themselves showed in terms of improvements in social behavior as assessed on performance-based tasks. One of the key findings was that we saw improvement on all three of those dimensions.
We’re now at the point where there’s potentially two [drugs], both targeting the same pathway.
The same issue of Science Translational Medicine where your study was published also reported findings from another clinical trial on vasopressin in autism, the VANILLA trial4. This study showed no improvement on the SRS following 12-week administration of a vasopressin antagonist. In your opinion, what are the reasons of the seemingly opposite results between your study and this other clinical trial?
It’s really interesting. Our primary outcome measure was positive; theirs wasn’t. But their secondary outcome measure, the Vineland-II Adaptive Behavior Scales, was positive. Given how heterogeneous autism is, it may be that there are some individuals who have increased vasopressin signaling, so an antagonist might work better for them, and it may be that there are individuals who have lower vasopressin signaling, who can benefit from a vasopressin agonist. But those studies just simply haven’t been conducted.
Are you going to follow up on these findings?
Right now, we have funding from the National Institutes of Health (NIH) to do a larger trial in a hundred kids (6–17 years of age), so we have expanded the age range, and they’re being dosed for longer. We would also like other scientists to replicate our findings independent of us completely. Another direction we’re taking is continuing our work on the monkey model. We’ve been giving intranasal vasopressin to low-social monkeys with low CSF vasopressin levels to test whether we can improve their performance on the laboratory tests that we developed with direct relevance to core autism features. We’ve also been able to identify infant monkeys that have subtle social information processing deficits that grow up to be low social.5 In the future, I’d like to see if we can treat these at-risk infants and test whether we can alter their social developmental trajectories if we intervene early enough.
What are the critical issues in autism clinical trial research today, in your opinion?
It was not feasible to perform spinal taps in kids before randomizing them in our first vasopressin treatment trial. It would be wonderful if we could noninvasively image vasopressin in the brain, right? We would be able to predict, in a very personalized way, who would respond to the vasopressin agonist, for instance, but we don’t have the technology to do that yet. There’s a lot of things that are critical to ensuring that we obtain the most accurate information from our trials. Having a study be completely double blind, right? Some people take that for granted, but many, many, many trials are not double blind. Using gold-standard clinical trial designs is really important. Having objective, well-verified endpoints in clinical trials that are reproducible is also critical, and I know the Simons Foundation had an RFA on this recently. The hope is that we will soon have measurement tools that are less subjective in nature as endpoints in clinical trials whether it be electroencephalogram (EEG) or eye tracking. We want to have more of these objective measurements in clinical trials to have a richer profile of what changes we might see. That’s some of what I would like to see moving forward in the clinical trials space.
Beside vasopressin, your research also focused on oxytocin in autism.
When we looked at spinal fluid levels of both vasopressin and oxytocin, we’ve always seen group differences in vasopressin levels in our low-social monkeys and in people with autism compared to controls1, 3, but oxytocin levels had been indistinguishable between experimental groups in both species.

Vasopressin seems to be a more important driver, or differentiator might be the better way to say it because we don’t know if it’s a driver yet, right? We published a paper a couple of years ago where we showed that people who had the lowest blood oxytocin levels responded the most to intranasal oxytocin therapy6. Maybe it’s possible that, because autism is highly heterogeneous, there’s a subset of people with oxytocin-signaling deficits who could benefit the most from this type of therapy. This is vast overgeneralization, but it may be that there are some individuals where their social deficits are more related to oxytocin-signaling deficits and there might be others where it may be more related to vasopressin deficits, and you’ll have other patients who could benefit from a GABAergic compound and others could benefit from a glutamatergic antagonist compound, right? The more that we can really rigorously profile the biology of individuals, that’s the way that we’re going to get traction for treating such a heterogeneous disorder.
Our goal is to diminish suffering and find treatments for individuals who can benefit.
You think back on cancer research thirty, forty years ago, we were treating everyone with chemotherapy or radiation. Now we have precision medicine treatments for cancer. I feel with the work that many in the field of autism are doing, we too are moving in this direction. If we understand the disorder biology better, we’ll be able to identify personalized treatments for people with neurodevelopmental conditions. For me, our goal is to diminish suffering and find treatments for individuals who can benefit.
In your opinion, what are the next scientific priorities in autism research?
My priorities are replicating our findings and really trying to move the field forward in terms of potentially having a medication that we can use. The other thing that our lab is working on is trying to identify not only biomarkers of people who are symptomatic, but also potentially people that may be at risk for developing autism. We know that the earlier kids receive behavioral therapy, the better their outcomes are. And we know we can diminish the lifetime cost of care by identifying kids early and treating them early. My lab is working on identifying a way to detect markers of autism in spinal fluid, or potentially other fluids, when people are symptomatic and also on identifying the disorder in infants who are at risk for developing autism before behavioral symptoms manifest. We can then get these children into treatment that much sooner.
References
- Parker K.J. et al. Sci. Transl. Med. 10 (2018) PubMed
- Parker K.J. et al. Sci. Transl. Med. 11 (2019) PubMed
- Otzan O. et al. Ann. Neurol. 84, 611-615 (2018) PubMed
- Bolognani F. et al. Sci. Transl. Med. 11 (2019) PubMed
- Sclafani V. et al. PLoS One 11, e0165401 (2016) PubMed
- Parker K.J. et al. Proc. Natl. Acad. Sci USA 114, 8119- 8124 (2017) PubMed


