
On May 15, 2018, behavioral, cognitive and computational neuroscientists convened at a workshop organized by SFARI on the so-called ‘excitation/inhibition (E/I) hypothesis’ of autism spectrum disorders (ASD). Originally proposed by John Rubenstein and Michael Merzenich in 20031, the E/I hypothesis postulates that an imbalance in excitation and inhibition disrupts the activity of neural circuits in individuals with the disorder.
This account has been highly influential in the field. Since its first formulation, however, significant advances have been made in the understanding of the genetic and neural bases of ASD. For instance, almost one hundred autism risk genes have now been identified with high confidence, and many others have been implicated at lower levels of confidence, animal systems based on several of those genes have been created and characterized, and the efficacy of pharmacological interventions targeting the inhibitory system (e.g., arbaclofen) are currently being examined.
One aim of the workshop was therefore to critically (re)evaluate Rubenstein and Merzenich’s E/I hypothesis in the light of the last 15 years of autism research. In particular, the workshop moderators, SFARI deputy director John Spiro and SFARI Investigator Bernardo Sabatini of Harvard University, drove the discussion around three main questions: Can the E/I hypothesis be articulated more precisely? What evidence supports an E/I imbalance in ASD? How does the E/I hypothesis inform the development of new therapeutic interventions for ASD?
Eleven speakers — Ofer Yizhar (Weizmann Institute of Science), Daniel Feldman (University of California, Berkeley), Gina Turrigiano (Brandeis University), Sacha Nelson (Brandeis University), Jessica Cardin (Yale University), Caroline Robertson (Dartmouth College), Timothy Roberts (Children’s Hospital of Philadelphia), Scott Murray (University of Washington), Alan Anticevic (Yale University), Dora Angelaki (Baylor College of Medicine and New York University) and Cian O’Donnell (University of Bristol) — presented their approaches to studying E/I balance from a number of perspectives, from cellular physiology, through neural circuits, to computational methodologies.
Charles Gilbert (The Rockefeller University), Peter Kind (Simons Initiative for the Developing Brain, The University of Edinburgh) and John Rubenstein (University of California, San Francisco) also joined the conversation as external discussants.
A summary of the topics discussed is presented below.
Homeostatic mechanisms
In its original formulation, the E/I hypothesis of autism predicted that, in a subset of individuals, overexcitation (i.e., increased ‘noise’) in ASD-relevant circuits might disrupt information processing and lead to abnormal patterns of activity (e.g., seizures). Several presentations discussed to what extent this formulation still holds and considered novel findings on E/I dynamics in mice harboring mutations in genes known to be strong risk factors for ASD and related neurodevelopmental disorders.
For example, SFARI investigator Ofer Yizhar showed that, consistent with the E/I hypothesis, mice with mutations in Cntnap2, a high-confidence autism risk gene, show increased cortical ‘noise’2. By recording neuronal activity in vivo, Yizhar found that neurons in the medial prefrontal cortex of Cntnap2 homozygous mice have enhanced activity compared with wild-type littermates. In the mutant mice, which are characterized by social deficits, these elevated levels of activity were also associated with a reduced capacity to learn and distinguish between social and nonsocial stimuli, thus suggesting a role of E/I imbalance in forming representations in behavioral domains relevant to ASD.
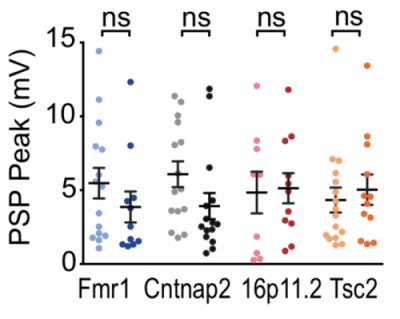
While studies have provided evidence for either increased excitation or reduced inhibition in ASD, the mechanisms driving those changes remain poorly understood. In one of the most comprehensive tests of the E/I hypothesis to date, SFARI investigator Dan Feldman discussed findings from a recent study in which he examined E/I ratios in four well-validated mouse models (Cntnap2, Fmr1 and Tsc2 knockout, and 16p11.2 deletion mice)3. Feldman demonstrated that all of these model systems were characterized by a marked reduction of inhibition and a smaller reduction of excitation in feed-forward circuits (layers 2-3) of the visual cortex, which led to an increase in the E/I ratio. At the circuit level, however, he did not find that increased E/I ratio translated into a greater firing rate, as predicted by the Rubenstein and Merzenich’s model (Figure 1). In fact, by modeling E/I dynamics, he reported that these changes served to keep synaptic activity stable. Feldman concluded that the increased E/I ratio observed in ASD may thus not constitute a primary deficit of the disorder but a compensatory response of homeostatic mechanisms.
That homeostatic mechanisms play a key role in regulating synaptic activity and account for changes in E/I balance was also emphasized by SFARI Investigator Gina Turrigiano, who studies plasticity of the mouse visual cortex. Driving this point even further, Sacha Nelson argued that homeostatic, compensatory mechanisms that typically re-stabilize activity after an insult may become maladaptive if they ‘overshoot’ to maintain optimal levels of information transmission4 (Figure 2). One outcome of this over-response is the propensity to develop seizures, which is often reported as a common comorbidity of ASD, as well as one of the strongest supporting arguments in the original formulation of the E/I hypothesis.
“It’s the dark side of homeostasis,” Nelson said.

As Turrigiano and Nelson pointed out, however, different homeostatic mechanisms can help stabilize neural activity and may be differently recruited in different cortical circuits. Which mechanisms may be implicated in ASD specifically and how they are regulated at the transcriptome level remains to be further investigated.
GABA
Key players in the E/I hypothesis are the excitatory (glutamate) and inhibitory (GABA) neurotransmitters of, respectively, pyramidal cells and interneurons. Presentations at the workshop mostly focused on interneurons and the GABAergic system, which have been widely implicated in ASD pathophysiology. GABAergic signaling pathways, and in particular, GABAB receptors, are also the target of arbaclofen, a potential drug treatment for ASD that has recently attracted the attention of the autism research community and Clinical Research Associates L.L.C, an affiliate of the Simons Foundation that has supported rodent- and human-based studies and provided the drug for ongoing clinical studies, such as the AIMS-2-Trials.
Starting with an analysis of the cellular components of the inhibitory system, SFARI Investigator Jessica Cardin argued that one reason why the E/I hypothesis of ASD is so complex and in need of more detailed specification is that interneurons are highly diverse in terms of their anatomy, physiology and different roles in neural microcircuits. Moreover, she argued that balance between E and I is represented in the temporal coordination between excitation and inhibition, as well as in synaptic balance, and that different classes of interneurons make different contributions to balanced activity. These observations suggest that the functional impact of different classes of interneurons on developmental processes (and ASD) should be considered and that current models assuming that E/I balance depends merely on the temporal coordination of pyramidal cells and interneurons may be too simplistic.
“There is need for more specificity,” Cardin argued.
In addition to a better understanding of the cellular and circuit mechanisms that underlie E/I balance, the workshop also highlighted the need for translational research in this area. To help bridge the gap between animal and human studies, SFARI Investigator Caroline Robertson described experiments that might capture E/I dynamics noninvasively in humans and that are potentially translatable to animal studies. The task that she described, known as ‘binocular rivalry,’ presents participants with two images, one per eye. As only one image at a time is consciously perceived, the paradigm measures the time the visual cortex ‘sees’ one image while suppressing the other —a process that is thought to engage E/I dynamics. Accordingly, Robertson uses this test as a marker of the strength of perceptual suppression in competitive excitatory and inhibitory interactions. She found that perceptual suppression by binocular rivalry is attenuated in individuals with ASD5 and shows no relationship with GABA levels, which positively correlated with the strength of perceptual suppression in the unaffected individuals instead6 (Figure 3). Building on these findings, Robertson will now examine the effects of GABAergic drugs on perceptual suppression through a SFARI Research Award that she was recently awarded.

In addition to the visual system, ASD has also been associated with deficits of inhibitory activity in the auditory domain. Reviewing his recent magnetoencephalography (MEG) and magnetic resonance spectroscopy (MRS) studies, SFARI investigator Timothy Roberts showed that the gamma-band activity (30-80 Hz) recorded from the superior temporal gyrus correlated with in vivo measurements of GABA concentrations in typically developing individuals, but not in children and adolescents with ASD7. As gamma oscillations are known to depend on inhibitory networks of GABAergic interneurons, Roberts argued that gamma-band coherence could serve as a biomarker and provide an objective measure to assess the effects of pharmacological interventions on E/I alterations in autism. With a SFARI Explorer Award, Roberts is currently examining the efficacy of arbaclofen, a GABAB agonist, on restoring gamma-band coherence and other electrophysiological measures that appear to be altered in autism.
E/I in autism and other disorders
As ASD is genetically and phenotypically heterogeneous, the conversation on E/I and human research also emphasized the need for the stratification of individuals participating in research studies and future trials. A case in point was provided by SFARI investigator Scott Murray, who discussed preliminary, unpublished functional magnetic resonance imaging (fMRI) findings showing both increases and decreases in neural excitability in the visual system in individuals with ASD. These results challenge a unidirectional understanding of E/I changes and call for a research approach that considers the neurobiological phenotype of ASD at a finer-grained level of analysis—something that would be particularly valuable to develop targeted interventions based on potentially different E/I profiles in ASD.
A final point of discussion in this session was the fact that alterations in E/I ratio are found not only in ASD, but also across a spectrum of other neurodevelopmental and psychiatric disorders. Alan Anticevic argued that pharmacological studies, neuroimaging methods and computational techniques should be combined to fully understand the core E/I mechanisms across disorders and synthetize different levels of analysis, from cells, through circuits, to behavior. Understanding the shared and distinct pathophysiological pathways of ASD and other brain disorders is a necessary step to help the field move forward, as studies have now shown that autism risk genes are implicated in a variety of conditions8 and that the molecular pathways of ASD and other disorders are partially shared9.
Computational approaches
While most presentations tackled the question of E/I imbalance through a bottom-up approach, others took a top-down view to investigate links between circuit-level abnormalities and behavior and to model the complex dimensions that account for E/I changes in ASD.
SFARI investigator Dora Angelaki, who uses neural networks and other computational techniques to study the effects of E/I changes on behavior, hypothesized that an E/I imbalance specifically alters nonlinear canonical computations in the brain of individuals with ASD10. By asking participants to solve tasks eliciting different aspects of multisensory perception that either rely on linear or nonlinear computations, Angelaki found that typically developing individuals and those with ASD similarly engage in linear operations such as multisensory integration of visual-vestibular and visual-auditory information; however, group-level differences arise in multisensory tasks involving nonlinear computations (i.e., casual inference — the capacity to integrate stimuli when they come from the same source).

Finally, SFARI investigator Cian O’Donnell described the formal models he has developed to assess the predictive value of the E/I hypothesis as originally formulated by Rubenstein and Merzenich11. By creating a circuit model of neurons in layers 2-3 of the mouse somatosensory cortex and testing in vivo data from a study of Fmr1 knockout
mice12, a model of fragile X syndrome, O’Donnell demonstrated that the original E/I model does not account for neuronal electrophysiological features other than firing rate. Instead, he demonstrated that a number of parameters modulated spike activity and that differences in E/I modulation existed also at different developmental ages (Figure 4). By showing that only a subset of the parameters modeling cell components had an effect on the input/output functions of the circuit, he also argued that one key challenge in studying E/I related processes is to identify which specific cellular alterations drive the behavioral symptoms in autism. His research has important implications for revisiting the E/I hypothesis and developing novel treatments, as it shows that E/I changes must be considered in the light of different developmental stages and a multidimensional model of circuit input/output function is needed.
Final remarks
Through the lens of circuit physiology, cognitive neuroscience and computational neuroscience, the workshop highlighted the deep influence of the E/I hypothesis in shaping the last 15 years of autism research. Not only has this account informed a mechanistic understanding of the disorder, but it has also inspired novel therapeutic avenues. The workshop also made apparent, however, that the original formulation of the E/I hypothesis may be too simplistic and fail to capture the complexity of the mechanisms driving an E/I imbalance in ASD. In particular, the field seems to lack a common, shared definition of what expressions like ‘E/I imbalance’ or ‘E/I ratio’ mean beyond the somewhat vague notion of altered ‘circuit excitability.’
“What we’ve learned today is that we can learn from each other, but we don’t speak the same language,” Sabatini said.
This lack of communication seems to be particularly evident when comparing E/I definitions in animal- and human-based studies, where researchers may use very different techniques (and tasks) to study the nervous system and behavior. While this variation may arise from the actual processes that can be examined in different species, there is a risk that “clear differences in E/I definitions can hinder discussions around translation,” noted Peter Kind, a discussant at the workshop.
Alongside a shared definition, the workshop also called for a definition of E/I that is specific to autism pathophysiology. E and I are common mechanisms that regulate circuit function (and dysfunction) and may therefore be involved in synaptic imbalance across a variety of psychiatric disorders. But in order to provide a meaningful understanding of autism and, potentially, inform therapeutic strategies, the E/I account should also provide a mechanistic account of the disorder.
“E/I balance can be used as a starting point for experiments to understand mechanisms, homeostatic or synapse/cell specific, but without deeper mechanistic insight, it should be interpreted with caution, as on its own, it may reveal little about the disorder being studied,” said Kind.
The field is also challenged by a dearth of translational studies, which makes it difficult to advance clinical research. From SFARI’s standpoint, developing a unifying framework will be important to promote communication and cross-fertilization between fields and approaches, as well as to inform the funding of projects based on the E/I hypothesis.
References
- Rubenstein J.L. & Merzenich M.M. Genes Brain Behav. 2, 255-267 (2003) PubMed
- Levy D.R. et al. bioRxiv (2018) Preprint
- Antoine M.W. et al. bioRxiv (2018) Preprint
- Nelson S.B. and Valakh V. Neuron 87, 684-698 (2015) PubMed
- Robertson C.E. et al. J. Neurosci. 33, 16983-16991 (2013) PubMed
- Robertson C.E. et al. Curr. Biol. 26, 80-85 (2016) PubMed
- Port R.G. et al. Autism Res. 10, 593-607 (2017) PubMed
- Buxbaum J.D. Biol. Psychiatry 77, 766-768 (2015) PubMed
- Gandal M.J. et al. Science 359, 693-697 (2018) PubMed
- Rosenberg A. et al. Proc. Natl. Acad. Sci. USA 112, 9158-9165 (2015) PubMed
- O’Donnell C. et al. Elife 6, e26724 (2017) PubMed
- Gonçalves J.T. et al. Nat. Neurosci. 16, 903-909 (2013) PubMed


