Despite the availability of whole-genome data sets in autism, identifying noncoding variants that confer risk of the disorder has been a substantial challenge for the field. Previous studies have suggested a role for de novo variants in predicted fetal brain promoters and regulatory regions (Turner et al., Am. J. Hum. Genet., 2016; Turner et al., Cell, 2017), paternally inherited structural variants in regulatory regions (Brandler et al., Science, 2018) and de novo variants in distal promoter regions (An et al., Science, 2018).
Now, Olga Troyanskaya, a Simons Foundation Flatiron Institute scientist, in collaboration with SFARI Investigator Robert Darnell, have identified a significant contribution of noncoding variation to autism risk at nucleotide resolution across the entire genome, at both the transcriptional and posttranscriptional levels.
In this new work, supported in part by a SFARI Research Award, the team applied a deep-learning-based framework to whole-genome sequences from 1,790 quartet families from the Simons Simplex Collection (SSC). This framework is informed at the DNA level by more than 2,000 genome-wide profiles of histone marks, transcription factor binding and chromatin accessibility, and at the RNA level by profiles of more than 230 interactions of RNA-binding proteins with RNA.
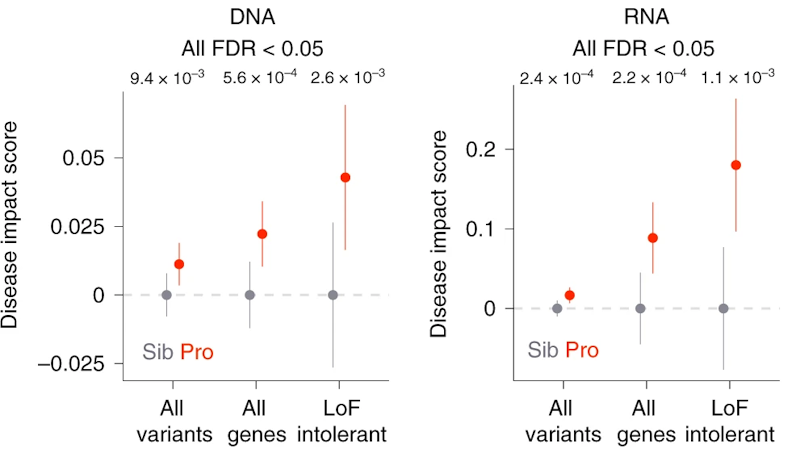
They found that a significant enrichment in probands for de novo noncoding mutations are predicted to have both transcriptional and posttranscriptional impact, as well as overall impact on disease. These variants converge on genes with (1) expression in the brain, (2) on genes involved in synapse function and chromatin biology, and (3) on genes previously implicated in ASD risk via coding mutations.
Importantly, the authors tested the functional impact of 59 of the putative mutations in reporter gene assays, finding that 57 of them show robust allele-specific effects on transcription. Finally, and of particular note, they report that individuals in the SSC at the lower end of the IQ spectrum have a higher burden of noncoding mutations with posttranscriptional effects, consistent with the notion that variants affecting mRNA splicing can have a strong impact on phenotype.
Reference(s)
Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk.
Zhou J., Park C.Y., Theesfeld C.L., Wong A.K., Yuan Y., Scheckel C., Fak J.J., Funk J., Yao K., Tajima Y., Packer A., Darnell R., Troyanskaya O.G.


