Over the last decade, mutations in CHD8 have been linked to autism spectrum disorder (ASD). A gene that encodes a chromatin remodeling factor, CHD8 has been shown to regulate a number of ASD-associated genes that are essential for brain and synapse development; however, the mechanisms by which changes in CHD8 may cause ASD remain unclear. A study in Chd8 haploinsufficient mice (Ellingford et al., Mol. Psychiatry, 2021) now finds that reduced Chd8 expression altered the balance of synaptic transmission in deep layer pyramidal neurons within the prefrontal cortex (PFC) — a specific cell type and brain region that have been implicated in ASD etiology.
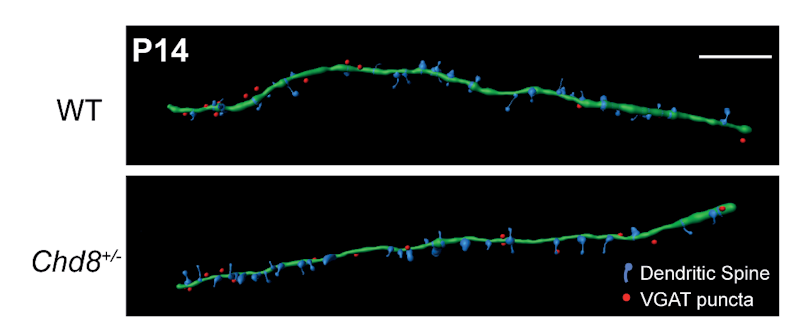
The work was supported in part by a SFARI Pilot Award and a Research Award to SFARI Investigators Laura Andreae and M. Albert Basson. Using a Chd8 heterozygous (Chd8+/–) mouse model, the researchers found that, compared to the wildtype, Chd8+/– mice had decreased excitatory and increased inhibitory synaptic transmission onto PFC deep layer pyramidal neurons, along with decreased firing. Interestingly, the most significant changes in this balance of excitatory/inhibitory transmission occurred during a key developmental window — the juvenile period (from postnatal days 14 to 20) — underscoring the importance of looking across different periods of neurodevelopment when studying synaptic transmission in ASD.
With further investigation, the researchers showed that the excitatory/inhibitory imbalance actually stemmed from distinct effects within individual subtypes of neurons. They found that Chd8 haploinsufficiency restricted to interneurons replicated the increase in inhibitory synaptic transmission seen in full Chd8 heterozygotes, without any changes to excitatory transmission. This indicated that the inhibitory transmission phenotype in Chd8+/– mice was driven entirely by a reduction of Cdh8 expression in interneurons, rather than through a global reduction across brain circuits.
On the other hand, when Chd8 haploinsufficiency was restricted to excitatory neurons, the effects replicated the decreased excitatory transmission seen in full Chd8 heterozygotes, but also displayed a complex activation of compensatory mechanisms in synaptic transmission. The fact that the full Chd8 heterozygotes did not display such compensation led the researchers to hypothesize that homeostatic mechanisms might be impaired in these animals. Indeed, the researchers next showed that neurons in Chd8+/– mice displayed a blunted ability to make compensatory changes in excitatory synaptic transmission and also showed inappropriate changes to inhibitory synaptic transmission. Andreae, Basson and colleagues ultimately proposed that dysregulation of homeostatic plasticity contributes to reduced excitatory/inhibitory balance in the PFC of Chd8+/– mice.
With these findings, the researchers offer a thorough characterization of a potential mechanism through which Chd8 mutations could impair the development and function of cortical circuits in ASD. The findings support the hypothesis that mutations in ASD risk genes converge on certain critical molecular pathways and cellular mechanisms to ultimately disrupt excitatory/inhibitory balance in ASD-associated brain circuits during neurodevelopment.
Reference(s)
Cell-type-specific synaptic imbalance and disrupted homeostatic plasticity in cortical circuits of ASD-associated Chd8 haploinsufficient mice
Ellingford R.A., Panasiuk M.J., de Meritens E.R., Shaunak R., Naybour L., Browne L., Basson M. A., Andreae L.


