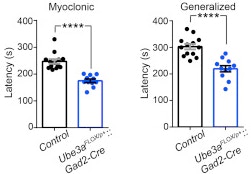
Angelman syndrome is a neurodevelopmental disorder resulting from the loss of the maternally inherited allele of UBE3A. It is characterized by severe developmental delay, motor dysfunction, profound speech impairment and seizures. SFARI Investigator Benjamin Philpot and his collaborators previously demonstrated that GABAergic inhibitory drive onto neocortical pyramidal neurons is greatly reduced in mice carrying a maternal UBE3A null mutation and that these deficits may contribute to the seizures observed in these animals (Wallace et al., 2012). Now, Philpot and his team have gone on to show that it is the specific loss of UBE3A from GABAergic, but not glutamatergic, neurons that causes enhanced seizure susceptibility. Surprisingly, this phenotype does not result from decreased GABAergic inhibition, but does correlate with increased accumulation of clathrin-coated vesicles within presynaptic terminals. Future studies focusing on the molecular pathways upstream and downstream of UBE3A within GABAergic neurons may help to uncover new strategies for treating seizures in individuals with this disorder.
Reference(s)
GABAergic neuron-specific loss of UBE3A causes Angelman syndrome-like EEG abnormalities and enhances seizure susceptibility.
Judson M.C., Wallace M., Sidorov M.S., Burette A.C., Gu B., van Woerden G.M., King I.F., Han J.E., Zylka M., Elgersma Y., Weinberg R.J., Philpot B.


