The International Angelman Syndrome Research Council (INSYNC-AS) held its first meeting virtually on July 9, 2021. A joint project of the Foundation for Angelman Syndrome Therapeutics (FAST) and the Simons Foundation Autism Research Initiative (SFARI), INSYNC-AS launched with the goal to drive acceleration of the translational research landscape related to treatments for Angelman syndrome (AS) and other neurodevelopmental disorders (NDDs). The meeting aimed to bring together experts with preclinical, clinical, regulatory and industry perspectives to discuss new ways to accelerate this initiative into successful therapies. These efforts were also aiming to form a foundation to serve as a model for other rare NDDs.
“The mounting knowledge and expertise for developing Angelman syndrome therapies could be leveraged for other neurodevelopmental conditions, which is something SFARI wants to expedite,” said John Spiro of SFARI.
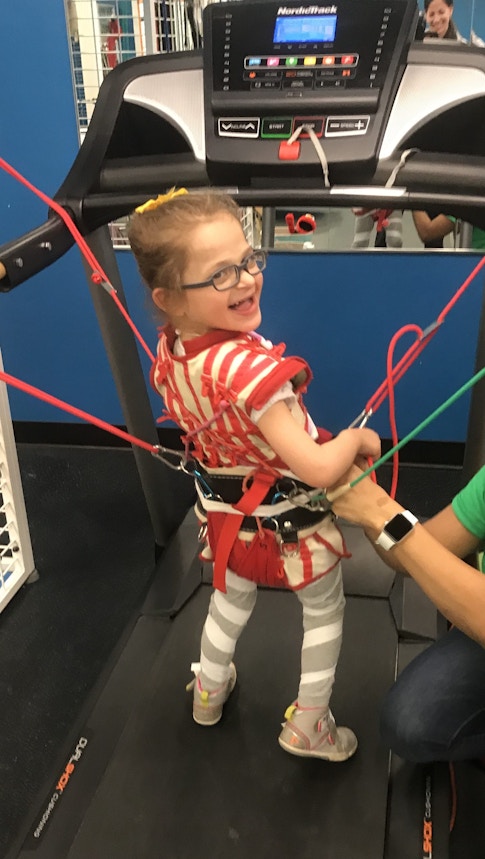
AS stems from a deletion, mutation or loss of function of the maternal copy of the UBE3A gene in neurons. UBE3A encodes an enzyme that ubiquitinates other proteins within neurons, tagging them for degradation. Without this, excessive by-products and proteins accumulate, which impedes synaptic function in neurons. People with AS face numerous challenges that prevent them from living independent lives: a universal lack of speech, debilitating seizures, severe developmental delays, difficulty walking and motor ataxia, fine motor dysfunction, sleep disturbances and feeding issues. Though AS is considered a rare disease, it is estimated to affect 1 in 15,000 people, which equates to about 500,000 people worldwide.
Allyson Berent, chief science officer for FAST and mother to Quincy, her 7-year-old daughter with AS, opened the meeting stressing the importance of working urgently and collaboratively to get therapeutic ideas hatched in the lab, through the regulatory process and, if appropriate, into clinical trials so they are eventually accessible to all affected individuals globally.
“INSYNC-AS created an opportunity for us to unite global experts to share the current landscape of translational research in Angelman syndrome,” said Berent. “Each of the council members in attendance were tremendously participatory and focused on the vision of the initiative to further translational science. The team was impressed with what has been accomplished in such a short period through FAST’s research initiatives and robust funding strategies.
Spiro and Jennifer Panagoulias, an aunt to a 19-year-old niece with AS and a regulatory expert who is supporting many AS focused initiatives, also made introductory comments, and encouraged participants to freely share their views in order to brainstorm the best pathways forward.
AS therapeutic landscape
As a co-discoverer of the genetic cause of AS, Arthur Beaudet of Luna Genetics gave his views on the prospects for therapies for AS and other NDDs. While optimistic about benefits for AS through gene therapy, he was more cautious for other NDDs given the seemingly irreversible brain changes that arise during disrupted fetal brain development. He argued that single gene disorders give the clearest targets, and advances in gene editing make it possible to correct a disrupted gene1.
The biology of UBE3A presents a unique opportunity for gene and disease-modifying therapies in AS. Normally, the maternal UBE3A copy is active in the brain, and the paternal copy is silenced; in AS, however, the maternal copy is missing or damaged, resulting in no or deficient UBE3A. Thus, re-activating the paternal copy of UBE3A in the brain, instead of replacing it, has become an attractive therapeutic approach for AS2.
This is complicated by the diverse genetics of AS. The most severely affected people carry a large deletion on chromosome 15 that removes not only UBE3A, but a number of other nearby genes, including some related to GABA inhibitory signaling; others have a missense mutation to UBE3A, or some have inherited two copies of the paternal UBE3A, leaving them without a working maternal copy in the brain3. To better understand the relationships between these genetic changes and pathogenicity, Beaudet called for a global database to compile AS-related genotypes and phenotypes.
Beaudet also outlined his view that inherited single gene conditions may be prevented through early prenatal diagnosis. This could involve genome sequencing of potential mothers and fathers prior to conception; if they are carriers of a disabling mutation, then genetic testing could be done on fetal cells found in the mother’s blood early in pregnancy.
In discussion guided by SFARI Investigator Matthew State (University of California San Francisco), participants agreed that AS is leading the way toward nucleotide-focused therapeutics and that even a partial correction of a genetic change could provide meaningful improvements. Other therapies aimed at targets downstream of the gene should not be discounted either, as combinations of therapies may work together to ameliorate various functional changes in NDDs.
Berent described the many activities FAST and the global AS research community have taken on to pursue all solid leads for AS therapeutics. FAST seeks to bring therapies to all genotypic versions of AS at any age to help all reach their full potential. Berent noted that evidence from individuals “mosaic” for the AS genotype (meaning some cells do not carry the AS-related genotype) suggest that even restoring a low level of the UBE3A protein to the brain, as low as 1–5 percent, may have transformational consequences; these people can speak words, walk well, and have very limited seizures, as well as a much less severe phenotype overall4.
Since its launch in 2008, FAST has become a nexus of information and support for AS research, bridging the interests of families of people with AS, academic researchers and industry. FAST has funded academic researchers to explore multiple approaches, with the idea of “de-risking” the AS research space to make it attractive for pharmaceutical companies. This includes funding research on developing novel animal models of AS, gene replacement therapies or multiple ways to reactivate the paternal UBE3A copy in the brain. FAST has also funded a core facility for centralized and rapid testing of potential therapeutics on various AS models. In 2017, FAST launched GeneTx Biotherapeutics to develop a novel antisense oligonucleotide to treat AS by unmuting the silenced paternal UBE3A gene. A phase 1/2 clinical trial of GTX-102 is ongoing.
FAST has also organized the community of parents and caregivers of people with AS in order to prepare the ground for clinical trials. This includes surveying over 300 parents to understand the domains in which they would most like to see improvements, as well as collaborate with other foundations to support the execution of a large natural history study through the National Institutes of Health (NIH) and the Food and Drug Administration (FDA) documenting the trajectory of symptoms of people with AS as they age. In 2016, FAST worked hand in hand with industry partners, other organizations and academic partners to establish the A-BOM (Angelman Biomarker and Outcome Measure) consortium, in order to pre-competitively develop biomarkers and outcome measures that are both meaningful, sensitive and appropriate for AS to be used in clinical trials. This information is essential for designing sensitive clinical trials based on the small numbers of participants with this rare disease, as well as to find therapies that actually make a meaningful difference in a person’s life. Berent is currently the director of this effort, and FAST has committed to invest up to $1 million per year to support this initiative, where 13 pharmaceutical companies are part of a pre-competitive steering committee driving priorities for endpoint development.
Discussant Mustafa Sahin of Boston Children’s Hospital, also a SFARI Investigator, noted that funding multiple approaches gives a higher chance of success. Others agreed, and added that FAST could be instrumental in managing family and industry expectations of the inevitable failures. A need for data sharing and standardization was also emphasized. With pharmaceutical companies involved, however, data is not always shared right away, and this can stall clinical trials or approvals. FAST has gotten around this somewhat by funding as much endpoint development as possible in a pre-competitive way so that all pilot data and measure can be shared. The team is working to create a mechanism in which companies can be more transparent in sharing placebo data, baseline data, as well as details on adverse events to help the entire field advance. The National Fragile X Syndrome Foundation helps promote openness and data-sharing by vetting pharmaceutical companies wanting to get involved in the fragile X syndrome space; if they are not endorsed by the foundation, then they cannot run trials in the clinics where most studies are conducted. Other foundations have also set up curated databases with standardized formats for sharing data. This has not yet been considered in the Angelman syndrome community.
Modeling AS
Finding ways to unmute the paternal UBE3A copy requires a detailed understanding of how it is silenced (“imprinted”) in the first place. Yong-Hui Jiang of Yale University described how these mechanisms might be revealed in studies of humanized mice that carry a human version of the UBE3A gene or in studies of cellular models of AS that are derived from each genotype of participant cells. Though these models only allow molecular, cellular and some physiological phenotypes to be studied, they serve as a basis for understanding paternal imprinting and the consequences of undoing it, as well as understanding how certain drug may impact different genotypes in different ways.
With support from FAST, Jiang is developing induced pluripotent stem cells (iPSCs) and brain organoids5 from people with AS to build a biorepository of all genotypes. Cell models from unaffected siblings are also being developed for comparison, and isogenic cell lines that share the same genome except for the AS-associated changes are also in the works. Jiang plans to have produced and characterized 20 cell lines in a year and will begin organoid production soon, as well as to set up a platform to allow distribution of these cell models to other researchers is the easiest and most pre-competitive way. Jiang is also developing a new mouse model of AS to demonstrate the impact of the haploinsufficient genes in the most common genotype for AS, the large 5–6Mb deletion, where 10–13 additional genes are also missing on the maternal allele. A mouse is being made to understand the phenotype differences when those 10–13 other genes are missing with, and without, Ube3a.
David Segal of the University of California, Davis gave a brief survey of animal models for AS. Though mice may not accurately predict whether a treatment will work in humans, Segal argued that mouse models of AS have taught the field a lot about AS biology and informed UBE3A reactivation treatment approaches. So far, most models consist of UBE3A maternal allele mutations, so these mouse models carrying a large deletion in the chromosome 15q11-q13 region is exciting.
FAST has funded the development of rat and pig models of AS, which could be better treatment models due to their robust phenotype, larger size, and that they are traditionally considered a more translatable species in drug development. Rat models that carry a full deletion of maternal UBE3A gene show large behavioral effects in vocalization, motor function and cognition6. Pigs that carry a UBE3A deletion have also been made; though not yet published, they are reported to show phenotypes related to the human features of AS, including hypotonia, motor deficits, reduced vocalization, delayed brain growth and seizures. The impact of these mutations on cognition has not been tested. Because pigs are large animals, they are well-suited to drug delivery or dosing experiments. Though safety and efficacy studies could be done in non-human primates, creating an AS monkey model would be difficult, incredibly expensive, take decades to have the mere numbers to be useful, and overall may be unnecessary.
In discussion, people agreed that large animal models such as the pig have an important role to play. Some worried that regulatory agencies would look to large animal models to provide efficacy data for a treatment, which could slow the pace of development. Others suggested that regulatory agencies could be persuaded to look to large animal models for understanding drug delivery and dosing, and not necessarily drug efficacy. At this point, it is too early to tell.
Delivering therapies
James Wilson of the Gene Therapy Program and Orphan Disease Center of the University of Pennsylvania described numerous potential delivery systems for gene therapy, including intravenous, direct brain injections, intrathecal, intraventricular and injections into the cisterna magna. Studies in larger animals, including non-human primates, find that intravenous delivery of adeno-associated virus (AAV) vectors do not cross the blood-brain barrier (BBB) very well7, whereas intrathecal or intra-cisterna magna (ICM) injections do reach the central nervous system8. This success is not without some safety concerns, however: dorsal root ganglion (DRG) neurons in the periphery seem highly prone to taking up the transgene, which can become a histopathologic adverse finding at high doses9. The translational impact of these results is still unclear clinically. Molecular methods to limit transgene expression in DRGs may help mitigate this finding10.
Beyond optimizing the route of delivery, researchers have also been working to develop viral vectors that more readily cross the BBB. For example, an AAV9 variant called PHP.B shows good BBB penetration after intravenous injection11, which suggests intravenous delivery of transgene-carrying vectors holds some promise. This biggest issue currently is that this capsid is only translatable in rodents and has not been successful to cross the BBB in primates. Indeed, many companies are working on finding similar BBB-crossing vectors for humans. During discussion, participants suggested that current delivery methods may be sufficient, knowing the data on mosaic individuals, where 1–5 percent of UBE3A expression could be transformative to over 97 percent of humans living with AS; researchers might better focus on excessive expression levels of UBE3A in the cells that have been transduced, perhaps through engineered promoters or other regulatory elements. However, the target levels of how many cells need to express the gene, and at what level within a neuron, still remain unclear for AS and is suspected to be lower than that initially considered.
Attracting industry
To understand how to attract drug companies to develop therapies for a rare condition like AS, Omar Khwaja of VectivBio described standard industry considerations when contemplating new therapeutic opportunities. They think about value, in whether there is opportunity to develop something transformative for a condition that has no disease-modifying therapy, and whether there are other companies competing in the same space; there is also a preference for developing therapies for a broad group of people, even in the sphere of rare diseases. They also think about how tractable a therapeutic idea is, weighing whether there are identifiable targets, understood mechanisms of action, established human in vitro models and animal models. They also consider how developable a therapy is, taking into account how easily it might be manufactured, whether there are regulatory precedents, and how well the clinical trial preparations have been made. Finally, there are also questions of whether going after a therapy is a good fit for the company at that time, which may depend on their current portfolio or finances.
In discussion led by Yael Weiss of Ultragenyx, participants pointed out that these are merely guidelines, and that many therapies pursued by companies do not tick all the boxes. Likewise, some innovative projects are overlooked when companies adhere too strictly to them. It was also noted that for rare diseases, competition from other companies is not necessarily a bad thing as it validates the approach. It is perhaps more difficult to be the first to bring something to market; once one is approved, this tends to spur investment to bring something different, or even better, as in the case of gene therapies for spinal muscular atrophy.
In her talk, Weiss then described how partnerships between foundations and industry can be fruitful, but that their respective goals do not always match. For example, though both may want to pursue diverse approaches, for foundations this will mean many therapeutic approaches for their focus disease, whereas for industry this may mean one approach for a rare disease that is one of several others in a disease portfolio. Likewise, foundations and industry will have different views on rights to a therapy. With these differences in mind, Ultragenyx has devised a bootcamp to help foundations attract interest from industry. This involves building a base of disease understanding and community, development of research tools, such as cell and animal models, and supporting research on an array of therapies. Weiss said that FAST-sponsored research activities have hit all possible therapeutic approaches, ranging from small molecules, to enzyme replacement therapy, to ASOs, to gene replacement therapy. This is serving as a model for nearly all other rare diseases in the space currently.
Questions arise as to how far research can be advanced by a foundation before it is passed onto an industry partnership. While foundations can quickly develop ideas from academia to provide proof-of-concept experiments, early partnerships with biotech can enable access to novel and useful platforms. Discussant James Wilson and others highlighted the difficulties in knowing when a project is ready to be handed over to industry. Even if the science is in place, the project is one of many in a company’s portfolio, and may stall for reasons not related to the science. This is frustrating to foundations, prompting some to steer the next stages of a project themselves, which FAST did in forming GeneTx Biotherapeutics to develop and commercialize the ASO GTX-102. This situation also highlights the importance of specifying what “due diligence” means in a partnership agreement: if an industry partner doesn’t advance a project by some pre-specified way or deadline, then rights may revert back to the foundation. As academia becomes more comfortable with tech transfer, smaller universities may be more open and agile to agreements with foundations than larger institutions currently are.
Clinical trial readiness
Jennifer Panagoulias focused on developing endpoints for clinical trials. As defined by the FDA, an endpoint must reflect something about how a person feels, functions or survives, whereas a biomarker indicates something about a biological process, such as a blood test or imaging. Panagoulias stressed that validated endpoints that have been used to gain regulatory approval in the past may not be appropriate for every disease. This means endpoints might need to be customized to the condition at hand, and foundations can provide valuable information gathered from families and caregivers to help formulate what a meaningful improvement might be. These disease-centered outcomes are increasingly being taken into account by the regulatory agencies.
As an example, Panagoulias told the story of the ORCA (Observer-Reported Communication Ability) measure developed specifically for AS through a collaboration between FAST and Duke University. This endpoint was inspired by FAST’s 2018 survey of parents or caregivers that found speech and communication to be the most important domain they would want to see improvement in. At the time, the standard speech/communication measure used as an endpoint in clinical trials did not appropriately measure receptive or pragmatic communication and was difficult to compare scores between participants. FDA recommended FAST develop a tool to capture the aspects of communication that are important to people with AS and their caregivers, such as conveying personal needs, like hunger. Collaborating with a team at Duke University, FAST created the ORCA tool, a caregiver-reported measure that includes expressive, receptive and pragmatic aspects of communication. The tool is sensitive to changes in communication ability over time in this population, doesn’t rely on speech and captures a range of abilities among people with AS avoiding a ceiling or floor effect that is so often seen on other measures in NDDs.
Measures that are sensitive to real world function are also increasingly desirable for regulatory approval, and digital health measures might also help capture some of these, such as sensors worn on the ankle that can monitor movement during the day while the person is at home. EEG recordings are also a biomarker of interest for AS, as they may reflect something about cognition and seizures. Biomarkers can sometimes be used as surrogate endpoints for diseases flagged by the FDA as needing accelerated development. Novel trial designs are also being explored that may be more appropriate for the small number of participants in rare disease trials12.
Discussant Elizabeth Berry-Kravis of Rush University Medical Center contrasted the approaches taken for AS to prepare for clinical trials with what happened in a failed clinical trials for fragile X syndrome: the trial used an endpoint already validated for regulatory approval, but which may not have been appropriate for the condition13. This stimulated a search for cognitive endpoints, which better capture core features of fragile X, as well as redesigns of fragile X clinical trials. This may have paid dividends: recent results from a phase-2 clinical trial that had safety and tolerability of a drug as primary endpoint for adults with fragile X found improvements among cognition and daily function measures included as secondary endpoints14. Berry-Kravis also argued that foundations could play an important role in educating the FDA about a condition and persuading them to accept novel endpoint measures that are more appropriate for a specific population.
Berry-Kravis is also involved in the currently halted clinical trial of the ASO GTX-102 for AS. She noted unexpectedly marked improvements in sleeping, walking and communication during the three months of ASO administration, and some of these were sustained after ASO delivery was halted.
The heterogeneity of the AS population (or any NDD) also came up in discussion. Heterogeneity may result in a failed clinical trial; if the treatment was beneficial to some participants but not to others, or if a drug is beneficial in certain domains in some people but different domains in others, then a drug can look ineffective, even though each person saw functional gains. Finding ways to stratify the small number of participants with rare disorders in controlled trials is challenging and so analyses that allow individuals to act as their own controls by looking at clinically meaningful change in trajectories pre- and post-treatment are a more sensitive way of identifying a drug effect. A dashboard of AS-specific endpoints was developed by the A-BOM consortium, and amongst them are some globally anchored assessments, like the Clinical Global Impression Scale for Angelman syndrome (CGI-I-AS), offering assessment across multiple domains and a somewhat individualized measure, but this still doesn’t address the conundrum that arises when a treatment works for some and not for others.
At the end of the meeting, Berent thanked people for their participation, and voiced hope to meet again in a year, in person. Spiro praised her energy and organization, and emphasized the urgent need to get safe drugs to help people with AS and other NDDs.
“The mission of FAST was the guiding light for the entire conference. It was clear by the end of the meeting that our mission was not only possible, but probable, and AS will serve as a model for so many other rare disorders where transformative clinical impact will be seen,” said Berent.
References
- Gillmore J.D. et al. N. Engl. J. Med. 385, 493-502 (2021) PubMed
- Meng L. et al. Nature 518, 409-412 (2015) PubMed
- Kalsner L. and Chamberlain S.J. Pediatr. Clin. North Am. 62, 587-606 (2015) PubMed
- Le Fevre A. et al. Am. J. Med. Genet. A. 173, 753-757 (2017) PubMed
- Sun A.X. et al. Science 366, 1486-1492 (2019) PubMed
- Berg E.L. et al. Transl. Psychiatry 10, 39 (2020) PubMed
- Ballon D.J. et al. Hum. Gene Ther. 31, 1237-1259 (2020) PubMed
- Hinderer C. et al. Mol. Ther. Methods Clin. Dev. 1, 14051 (2014) PubMed
- Hordeaux J. et al. Hum. Gene Ther. 31, 808-818 (2020) PubMed
- Hordeaux J. et al. Sci. Transl. Med. 12, eaba9188 (2020) PubMed
- Chan K.Y. et al. Nat. Neurosci. 20, 1172-1179 (2017) PubMed
- Lake S.L. et al. Stat. Med. 40, 4167-4184 (2021) PubMed
- Berry-Kravis E.M. et al. Sci. Transl. Med. 4, 152ra127 (2012) PubMed
- Berry-Kravis E.M. et al. Nat. Med. 27, 862-870 (2021) PubMed
- From parent advocate to nonprofit chief science officer, to biotherapeutic company cofounder — A personal journey through drug development for Angelman syndrome
- Small molecules, genes and antisense oligonucleotides: Industry perspectives on treatment development for ASD
- Clinical trials and cyclic AMP in fragile X syndrome: A life journey
- SFARI workshop explores challenges and opportunities of gene therapies for autism spectrum disorder


