Although fragile X syndrome (FXS) has been associated with changes in synaptic development, increased mRNA translation and mitochondrial dysfunction, the precise relationships between these features of the disorder have been unclear. A new study provides critical mechanistic insight into the mitochondrial abnormalities in FXS in mice and humans, their link to synaptic function and their ultimate importance in driving the associated behaviors.
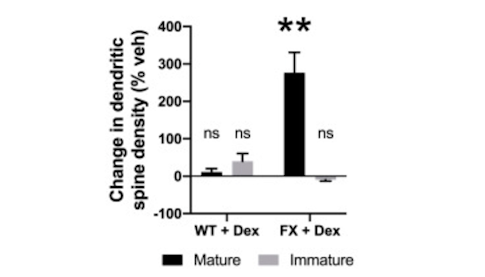
The work was supported in part by a Pilot Award to SFARI investigator Elizabeth Jonas, who led a group using both FXS fibroblasts and Fmr1-deficient mouse neurons to identify a mitochondrial inner membrane leak as a key driver of neuronal and behavioral abnormalities in the syndrome. This leak, which results in inefficient ATP synthesis, stems from an imbalance in the production of the beta- and c-subunits of the mitochondrial ATP synthase. Specifically, they showed that the fragile X protein is required for the stimulus-induced phosphorylation of the translation factor EF2, which promotes both translation of the beta-subunit and a transition from a ‘leak’ phenotype toward oxidative phosphorylation. In the context of FXS, Jonas and colleagues showed that depletion of the c-subunit also has the effect of establishing the appropriate ratio of ATP synthase enzyme subunits, closing the leak channel and, in turn, triggering synapse maturation. Of note, closure of the ATP synthase leak by administration of dexpramipexole to 2-month-old Fmr1-deficient mice resulted in a reduction of repetitive behaviors and hyperactivity.
The new results may prompt investigators to ask whether deficits in mitochondrial function, leading to altered synaptic maturation, also contribute to idiopathic forms of ASD.
Reference(s)
ATP synthase c-subunit leak causes aberrant cellular metabolism in fragile X syndrome.
Licznerski P., Park H.-A., Rolyan H., Chen R., Mnatsakanyan N., Miranda P., Graham M., Wu J., Cruz-Reyes N., Mehta N., Sohail S., Salcedo J., Song E., Effman C., Effman S., Brandao L., Xu G.N., Braker A., Gribkoff V.K., Jonas E., Valdivia Espino J.


