I’ve been reading “The Serengeti Rules1,” the excellent new book by scientist and educator Sean Carroll. In it he provides a highly readable survey of the features that many biological systems hold in common. These range from the molecular details of bacterial enzyme induction to ionic balance in the bloodstream to the large-scale interactions between species on the Serengeti Plain. The linchpin at every organizational level of life, he argues, is homeostatic regulation — the ability of a cell or an organism or an ecological unit to use feedback to maintain key parameters within an acceptable range. Conceptually, the reason the number of wildebeests on the Serengeti is what it is turns out to be not so different from the reason blood pH is typically maintained within certain limits. Carroll concludes with a sensible plea for us to understand these regulatory rules so that we can better protect the planet from the effects of environmental degradation and climate change. But his argument has stayed with me for another reason.
Numerous reviews on the neurobiology of autism have also focused on the idea of homeostasis as a way to better understand a body of literature that is vast and often difficult to synthesize. Here, I will mention a couple of recent reviews on this topic. I will also discuss several other reviews that provide interesting and complementary overviews of recent progress in understanding the genetic architecture of autism and the potential importance of deficits in neural progenitor cell proliferation and differentiation in the development of the disorder. Together, these reviews clearly show the extent to which basic research on autism has grown over the past decade. By coming to grips with the diversity of papers out there, these reviews provide a service to the autism research community, not least by succinctly summarizing key ideas in the many valuable figures and tables contained within their pages, which will no doubt be referred to for many years to come.
Autism risk genes and synaptic homeostasis
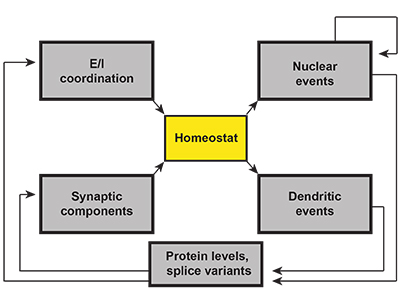
Richard Tsien and his colleagues’ comprehensive review earlier this year focuses on the idea of synaptic homeostasis as a possible unifying mechanism in autism2. Although this idea is not a new one3, the current version takes advantage of the knowledge gained about particular autism risk genes to make the fullest possible case for it. Rather than classify risk genes according to cell types or regions of the brain in which they’re expressed, the authors map gene functions on to what they call a ‘canonical homeostatic feedback loop.’ This loop features a critical sensor, or ‘homeostat,’ which regulates the synaptic properties of a neuron (Figure 1). Any alteration of the feedback to the sensor (i.e., any mutation in a gene encoding a component of the feedback loop) might result in the same outcome, whether it occurs in a transcription factor, an ion channel or a structural component of the neuron.
Thomas Bourgeron discusses the genetic architecture of autism with a similar view in mind4, by arguing that mutations that disrupt synaptic homeostasis might be well or poorly tolerated depending on the genetic buffering capacity of the individual. In other words, particular genetic backgrounds may be more vulnerable to mutations (or environmental factors) that perturb neural function through the modulation of synaptic strength. It’s worth noting that Graeme Davis’ SFARI-funded project is designed to experimentally test ideas along these lines (using genetic interaction screens in Drosophila).
Bourgeron’s review also includes a simplified schematic that illustrates how autism risk is likely associated with the presence or absence of a modest number of rare alleles acting in the context of a burden of common susceptibility alleles that exists as a continuum in the population (Figure 2). This viewpoint is shared by Dan Geschwind and his colleagues in their recent review article5. Indeed, such a view may represent a general consensus in the field; however, it is often obscured by individual papers that focus entirely on one particular type of risk variant to the exclusion of all others.
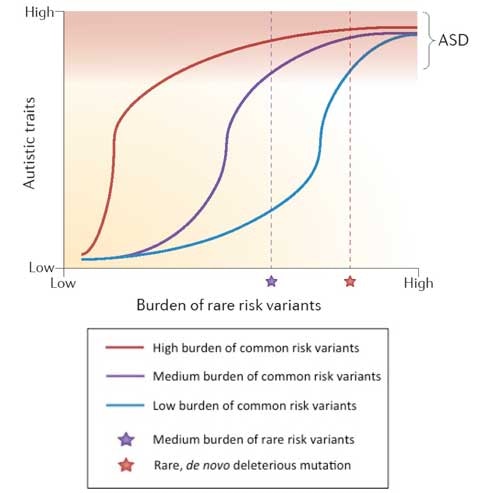
Geschwind’s review also features two strikingly effective figures — one representing a comprehensive view of the genetic architecture of autism and another summarizing what we know about the convergent neurobiology of the disorder at the molecular, cellular and circuit levels5. It is also worth noting that of the 300 citations in the review by Geschwind and his colleagues, only 6 predate the year 2000, reflecting increases in public and private funding over the past 15 years and the intensity of our combined efforts to understand the origins of autism in the developing brain.
Autism risk genes and progenitor cell biology
Autism risk genes and progenitor cell biology
Three other reviews — authored by Carl Ernst6, Gaurav Kaushik and Konstantinos Zarbalis7, and my own review8 — are also noteworthy because they were all published around the same time and converge on a common theme: that a large number of autism risk genes have roles in prenatal neurogenesis. Specifically, based on a number of studies of mouse knockouts, many of these gene products were found to regulate the proliferation of neural progenitors or the migration and differentiation of neurons in the cortical plate (Figure 3A, 3B). These mouse studies are consistent with data from human neuropathology, gene expression and emerging studies of induced pluripotent stem cells (iPSCs) derived from individuals with autism. As each month passes, additional autism-related risk factors are associated with disruptions of these same populations of cells, including, for example, long non-coding RNAs9 and bacterial infection during pregnancy10. Taken together, with similar arguments made in the review by Geschwind and his colleagues5, this collection of reviews suggests that at least some of the initial disruptions of brain development in autism may occur well before synaptogenesis begins (Figure 3C).
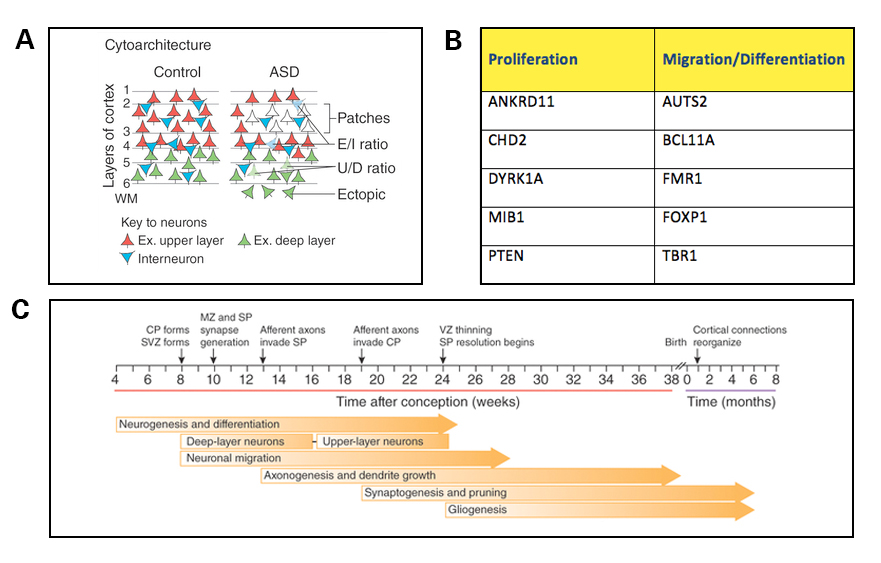
A) Altered cortical cytoarchitecture has been observed in mouse models of autism and human postmortem tissue from individuals with autism. Such changes may arise from combined deficits in neural proliferation, differentiation and migration during fetal development and alterations in synaptic function. Cortical layers are indicated by numbers. E/I, excitatory or inhibitory neuron; Ex. upper layer, excitatory upper layer neurons; Ex. deep layer, excitatory deep layer neurons; U/D, upper-layer or deep-layer neuron; WM, white matter.
B) A large number of autism-risk genes are involved in neural proliferation, migration and differentiation during early cortical development. Examples of a few of these genes are listed here. For additional risk genes in these categories, please refer to the following reviews: Ernst C.6, Kaushik G. and Zarbalis K.7, Packer A.5
C) Timeline of human brain development. Neurogenesis, differentiation and migration begin in the first trimester of gestation, and synaptogenesis begins at 18 to 20 weeks post-conception. CP, cortical plate; MZ, marginal zone; SP, subplate; SVZ, subventricular zone; VZ, ventricular zone. Panels A and C are reproduced from de la Torre-Ubieta L. et al5.
Relationships between early developmental effects and synaptic and network dysfunction
How might these reviews shape the direction of the field? One central issue that some of them grapple with is the relationship between early developmental effects and later synaptic and network dysfunction. It may be that autism risk genes have independent and important effects on both early developmental events and on synaptic function. For example, components of the autism-associated BAF complex of chromatin remodeling proteins likely have roles both in progenitor cell biology and in synapse and dendrite morphology and function, and together these effects could result in autism phenotypes11,12. Alternatively, the behavioral effects of mutations on presynaptic biology may be largely dependent on the ultimate effects these developmental changes have on synaptic function. As Ernst points out in his review6, understanding these relationships may help to determine to what extent autism-related phenotypes can be treated with interventions that are administered postnatally.
Taken together, these recent reviews provide a helpful framework as autism researchers try to convert new genetic insights into a better understanding of the neurobiology of autism. Although new tools such as human-derived iPSCs and organoids will enable experiments that avoid the drawbacks of the current range of animal models, a difficult question remains: What, exactly, should one assay for? The overviews discussed here make a good case for assessing the effects of autism-associated mutations on basic cellular attributes such as cell cycle length, cell migration and cell differentiation, with a long-term goal of understanding whether and how variation in these parameters affects the ability of neurons in relevant circuits to maintain synaptic activity within a normal range.
References:
- Carroll S.B. (2016) The Serengeti rules: The quest to discover how life works and why it matters. Princeton, New Jersey: Princeton University Press.
- Mullins C. et al. Neuron 89, 1131-1156 (2016) PubMed
- Ramocki M.B. and H.Y. Zoghbi Nature 455, 912-918 (2008) PubMed
- Bourgeron T. Nat. Rev. Neurosci. 16, 551-563 (2015) PubMed
- de la Torre-Ubieta L. et al. Nat. Med. 22, 345-361 (2016) PubMed
- Ernst C. Trends Neurosci. 39, 290-299 (2016) PubMed
- Kaushik G. and K.S. Zarbalis Front. Chem. 4, 12 (2016) PubMed
- Packer A. Neurosci. Biobehav. Rev. 64, 185-195 (2016) PubMed
- Rani N. et al. Neuron 90, 1-15 (2016) PubMed
- Humann J. et al. Cell Host Microbe 19, 388-399 (2016) PubMed
- Zhang Z. et al. Mol. Cell. Biol. 36, 70-83 (2015) PubMed
- Ka M. et al. J. Neurosci. 36, 2723-2742 (2016) PubMed