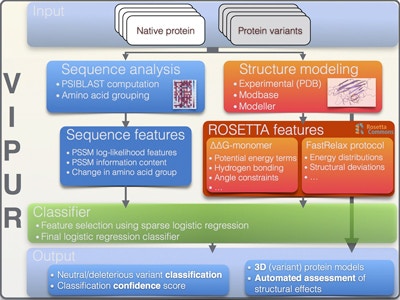
VIPUR analysis pipeline: Starting from a structural model of the native protein and a list of variants to be tested, VIPUR identifies and interprets deleterious variants. Image from Baugh E.H. et al. (2016).With the increasing availability of human genomic data, researchers have a wealth of information that they can mine for disease susceptibility variants. The majority of existing variant annotation methods use only sequence-based or structure-based information to predict whether a variant is likely to be deleterious or neutral. Richard Bonneau (a Simons Center for Data Analysis scientist) and his colleagues have now developed a novel computational framework, VIPUR (Variant Interpretation and Prediction Using Rosetta), which combines sequence analysis and protein structural modelling to identify, rank and interpret deleterious variants. In a proof-of-concept study, the researchers tested VIPUR’s ability to identify autism risk variants by classifying de novo missense mutations in the Simons Simplex Collection (SSC). This method predicted 43 variants (out of a total of 2,226 variants) that likely result in disrupted protein function and merit further investigation.
Reference(s)
Robust classification of protein variation using structural modelling and large-scale data integration.
Baugh E.H., Simmons-Edler R., Müller C.L., Alford R.F., Volfovsky N., Bonneau R.