
Information gathered from our senses is critical for the development of social behaviors and communication skills. Touch perception has been shown to be particularly important during the early developmental years, with studies in both animal models and human infants suggesting that a lack of nurturing touch can lead to later deficits in social behaviors.
The majority of individuals with autism spectrum disorder (ASD) present with hyper- or hyposensory sensitivities, including touch sensitivities. SFARI Investigator David Ginty, with help from a 2015 SFARI Pilot Award and a more recent SFARI Research Award in 2017, has been using mouse models to assess how ASD-risk genes affect touch sensation and to understand what effects these sensory alterations might have on the social deficits seen in ASD.
I recently caught up with Ginty to talk about his ongoing work on the links between ASD risk genes, sensory neuron function and social behaviors, and to get his take on the importance of these findings for the ASD community. The interview has been edited for clarity and brevity.
Your laboratory has been interested in understanding the development, organization and function of the neural pathways involved in the sense of touch for many years. What got you interested in studying the role of touch sensation in autism?
Clinical and anecdotal observations suggest that individuals with autism, and in particular children with autism in particular, are often aversive to what we would normally consider innocuous touch stimuli. It’s an exciting time in the somatosensory field, because we’re gaining a much greater understanding of how the peripheral sensory neurons of touch respond to tactile stimuli and how the touch information that these sensory neurons extract from the physical world is conveyed to, represented and processed in the central nervous system to underlie perception. Because of this increased understanding and a range of new, tremendously powerful tools for studying and understanding the somatosensory system that have been developed in the last years, including mouse models with high construct validity for autism spectrum disorder, we began to think that we were poised to understand what components of somatosensation go awry in autism. We were able to ask whether somatosensory alterations can be recapitulated in mouse models of autism spectrum disorders and, if so, which aspect of the somatosensory system is dysfunctional. Ultimately, we could ask what the contributions of this sensory deficit might be to other core features of autism and whether understanding sensory processing deficits in ASD might offer clues for early diagnosis or approaches for treating tactile deficiencies and perhaps other aspects of the disorder.
We found that the loss of the presynaptic inhibition brake in the peripheral nervous system of these autism mouse models was also tightly associated with anxiety-like behaviors, which are indeed often co-morbid with autism.
In 2016, your laboratory published findings relating autism risk genes to deficits in peripheral touch neurons. What were the main findings that came out of that work?

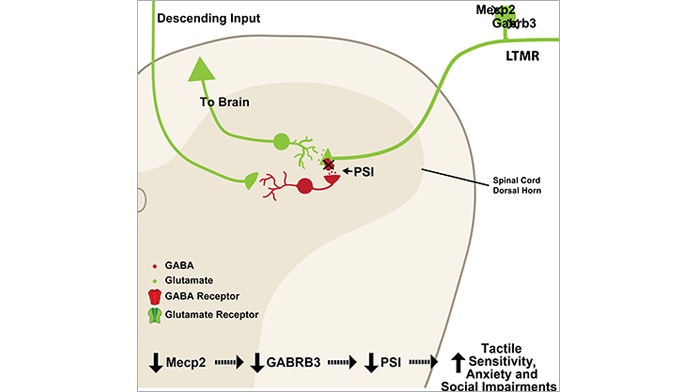
This work1 was spearheaded by Lauren Orefice, an exceptionally talented postdoctoral fellow in the lab. Orefice first found that different several mouse models of autism are, in fact, hypersensitive to innocuous touch. Interestingly, she saw tactile hypersensitivity and a deficit in tactile acuity when MECP2 — which is responsible for Rett syndrome, and GABRB3, which is associated with non-syndromic forms of autism — were deleted selectively in peripheral sensory neurons. This suggests that altered touch sensation in autism may be due to peripheral, instead of brain-based, deficits.
Orefice and her coworkers in the lab then showed a mechanism for the tactile deficits in these two mouse models. Normally, the flow of sensory information from the periphery to the central nervous system is conveyed by the primary sensory neurons that synapse onto second-order neurons located in the spinal cord and brainstem; these neurons then convey touch information to higher brain regions. That synapse is subject to what we call presynaptic inhibition, which normally acts as a ‘brake’ on the system. We found that this presynaptic inhibition brake was greatly diminished in both mouse models, so the flow of sensory information from the periphery into the central nervous system was greatly augmented. This is the locus of dysfunction that underlies tactile hypersensitivity in the MECP2 and GABRB3 mutant mouse models.
But perhaps most surprising, we found that the loss of the presynaptic inhibition brake in the peripheral nervous system of these autism mouse models was also tightly associated with anxiety-like behaviors, which are indeed often co-morbid with autism. This finding makes an interesting connection between sensory experience, tactile hypersensitivity and anxiety. Crucially, we saw this connection only when there was a developmental alteration in tactile sensitivity. This is fascinating because it shows a link between the development of normal touch sensitivity and the development of anxiety-like behaviors. We are very interested in further exploring why that is the case.
I think that, while genetic alterations clearly manifest at the level of the brain, we have to consider deficits at the subcortical level, including the spinal cord and peripheral nervous system.
David Ginty Laboratory / Harvard University
You mention that these results were, at least initially, somewhat surprising to you. What sorts of reactions has the research community expressed to this idea that alterations in peripheral touch would underlie autism-like behaviors?
The vast majority of, if not all, individuals with autism present with a degree of over-reactivity to one or multiple sensory modalities. The reaction we’ve received from the community is that it makes sense that sensory overload is associated with, and perhaps a causal or contributing factor for, at least a subset of the core autistic-like phenotypes. While surprised initially, many scientists and non-scientists seem to have embraced the idea that there is likely a functional relationship between sensory overload and sensory processing deficits as well as other behavioral deficits such as anxiety and altered social interactions.
This published work points to two specific neurodevelopmental-risk genes, Mecp2 and Gabrb3, as playing a role in touch-mediated alterations in social behaviors. Do you think that this is likely to be a more general mechanism caused by autism risk genes?
In Orefice’s 2016 paper, we used a mouse line that allowed us to delete MECP2 in virtually all of the cells below the neck but leaving MECP2 intact in the head and neck, including the brain. To our surprise, we found that most of the overt deficits — abnormal breathing, motor coordination, sensory deficits, the shortened life span and hind-limb clasping deficits — seen in whole animal MECP2 knock-out mice were recapitulated in this below-the-neck animal model. What that strongly suggests is that a lot of what is wrong, at least in Rett syndrome, is because of that gene being required not in the brain but in other parts of the body, including the nervous system below the neck.
The vast majority of work in autism is focused on trying to understand the function of risk genes in the brain. But I think that, while genetic alterations clearly manifest at the level of the brain, we have to consider deficits at the subcortical level, including the spinal cord and peripheral nervous system. It is also interesting to consider how peripheral sensory information processing deficits lead to alterations in brain development. Of course, we’ve known for more than 50 years that sensory experiences play a crucial role in brain development. How sensory overload during early life affects the developing brain has now become a very compelling question.
We have a lot of ongoing work addressing the parts of the nervous system that other autism-risk genes might be affecting. We’re using the principles we’ve learned from the MECP2 and GABRB3 study to address how widespread this is in terms of autism-associated gene function in the periphery.
David Ginty Laboratory / Harvard University
Your 2016 paper specifically related developmental alterations in touch perception to anxiety-like behaviors. How does this developmental effect change the way we think about these types of sensory deficits in ASD?
To answer this question, we would need to know at what stage, developmentally, perhaps even in utero, somatosensory overload is occurring and contributing to brain and behavioral deficits. One challenge is that we don’t know how developmental milestones and ages in a rodent model that would be comparable to the precise developmental stages of humans. We also need more quantitative approaches, and access to affected individuals at different ages would be critical to understand when and in which patient populations such deficits are most pronounced.
To this end, Orefice and I have now initiated a collaboration with Alvero Pasqual-Leone (Beth Israel Deaconess Medical Center, Harvard) and Alex Rotenberg (Boston Children’s Hospital) to look at tactile sensitivity in individuals who have either syndromic or idiopathic forms of autism. Here, we are asking whether the same tactile hypersensitivities that we see in the mouse models are present in children with developmental disorders, including autism. Is one patient population particularly sensitive compared to another? We’re using some of the same tactile sensitivity assays developed for the mouse work for measurements in humans. This work will allow us to measure the extent of tactile hypersensitivity in genetically defined autism populations and, when it exists, whether there are particular populations that are more susceptible.
The long-term goal of this project is to begin to think of ways of testing potential therapeutic interventions and seeing if alleviating hypersensitivity early on has any impact on the development of autism-related behaviors, for example, on anxiety.
I’d love to see relationships between basic scientists like myself and clinical scientists be supported more.
Beyond your own work, what steps would you like to see the research and clinical communities taking to help to move these findings from model systems into a better understanding of the human condition?
I’d like to see similar questions being asked in all sensory domains, including taste, as well as in other autism model systems; I’d like to see connections being made between sensory overload and different overt behaviors. For example, is there a tactile component to the averseness some individuals with autism show towards particular foods?
On the therapeutic front, we should explore the relationship between what we see in animal models and what we see in humans. I’d like to see a more quantitative approach to measuring sensory responsiveness in different cohorts of individuals with autism and, in particular, knowing when these deficits emerge. Some of this type of work is now beginning for the sense of touch. If sensory overload happens early and with a particular signature, it will become interesting to ask whether sensory measurements could be used for earlier diagnoses or as biomarker for drug efficacy.
You recently received additional funding from SFARI to continue these research interests. Do you see other ways that SFARI might help to push this research forward?
Our work really wouldn’t be happening without the support of SFARI. In fact, SFARI has been hugely instrumental for moving this entire field forward, and I applaud the SFARI leadership for their emphasis on both basic and translational work in their focus and funding mechanisms, and for helping to bridge basic and clinical research.
In the future, I’d love to see relationships between basic scientists like myself and clinical scientists be supported more. There is great value in having conferences where both clinical and basic scientists meet and discuss basic mechanisms and therapeutic opportunities. To really translate findings like the ones we have, there is a need to support collaborative interactions between basic scientists, who probe mechanisms in model systems, and clinical scientists, who have great expertise in the human condition and direct access to autism populations. I think that SFARI is uniquely positioned to help scientists who study basic mechanisms rub elbows with physicians and physician-scientists who evaluate and treat individuals with these disorders, so that together we can develop new ways of treating them.


