
PDF documents: Announcement | FAQs
SFARI initiated whole-genome sequencing (WGS) of the Simons Simplex Collection (SSC) in early 2015. This update is intended to inform the research community about the latest data that are now available, as well as to outline future sequencing plans for the collection.
Current data availability
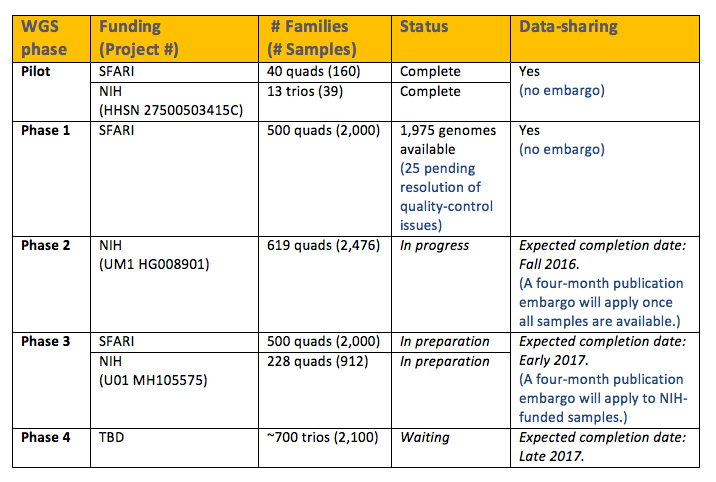
A total of 2,174 whole genomes are currently available. This includes samples from the Pilot and Phase 1 batches (see Table 1).
The Pilot study includes a total of 199 genomes: 160 genomes from 40 quad families (defined as both biological parents, the affected child and an unaffected designated sibling) and 39 genomes from 13 trios. An initial analysis of these samples has recently been published1.
Phase 1 includes a total of 2,000 genomes (from 500 quad families). The majority of these genomes (1,975 genomes) are now available; the remaining 25 genomes are expected to be available shortly, pending resolution of some quality control issues.
* Note: All of the genomes outlined above are available to the research community with no publication embargo restrictions.
Future plans
Plans are underway to perform WGS of the entire SSC. That effort is made possible by funding from SFARI and the National Institutes of Health (NIH).
Table 1 outlines the expected timeline (and data-sharing agreements) for the sequencing of the next batches of the SSC. Sequencing of all quad families in the SSC is expected to be completed in early 2017 (Phases 2 and 3). The remaining families (about 700 trios) will be sequenced in late 2017 (Phase 4).
We will update the SFARI website as soon as new data batches are released.
**Note: A four-month publication embargo will be imposed for the two batches of samples being sequenced under NIH funding, to allow for the principal investigators of those grants to publish an initial analysis.
Which researchers can use the WGS data?
WGS data are available for use by the entire research community, regardless of SFARI funding, with no restrictions other than the following:
- The intended research must be relevant to autism and/or other neurodevelopmental disorders, and
- researchers must agree to abide by any publication embargos that apply.
Genome sequencing details
All WGS data are generated from whole-blood DNA. All genomes are being sequenced at the New York Genome Center (with the exception of 13 trio families in the Pilot study, which were sequenced at the University of Washington). Additional details are available in the FAQ document.
All sequencing is via paired-end reads (2 x 150 bp) to 30x mean coverage and will be available in BAM file format containing all passed filter reads and quality scores.
The Pilot and Phase 1 alignment of reads to hg19/NCBI build 37 was done using Burrows-Wheeler Aligner (BWA) software package (BWA-MEM algorithm), and all local realignment and variant calling was done using Genome Analysis Toolkit (GATK) best practices (version 3.4).
Phase 2 and 3 sequences may be analyzed using a different computational pipeline, which will be announced as soon as details are finalized. Pilot and Phase 1 sequences will then be re-analyzed using this new approach to allow for direct comparisons between all families.
In addition to BAM files, single nucleotide variant (SNV), indel and structural variant calls are available.
How to access the data
All of the data will be made available by request after logging into SFARI Base and completing an application. See here for more information about the application process.
Once the application is approved, the data can be accessed via the following three options:
- Cloud-based access (via Amazon Web Services)
- Fermilab access (data can be transferred to research institute servers via GridFTP)
- Simons Foundation server (data can be transferred to research institute servers via globus.org)
Additional details about how to download or access the data (estimated to be roughly 300 TB for the Pilot and Phase 1 data) will be provided to researchers after their SFARI Base application has been approved.
Additional information
- An FAQ document with detailed information about sample selection and data access is available here.
- Additional information about the SSC (including a list of publications that have used SSC genomic data) is available here.


