
Smith-Magenis syndrome (SMS) is a rare developmental disorder characterized by a diverse set of symptoms, including intellectual disability and delayed speech and language skills. The laboratory of SFARI Investigator Liqun Luo (professor, Stanford University, and Howard Hughes Medical Institute Investigator) has traditionally been known for its studies of how neural circuits assemble during development and for the development of novel genetic tools. With a push from two talented individuals, postdoctoral fellow Wei-Hsiang Huang and graduate student Casey Guenthner, Luo’s laboratory is now also studying developmental disorders. Luo’s team published a paper in 2016 that dissected the molecular and neural functions of RAI1, the causal gene for SMS1.
I recently spoke with Luo to discuss this paper, to hear his thoughts on the field and to find out his laboratory’s future plans for studying SMS. The interview has been edited for clarity and brevity.
Could you start by giving us some background on SMS?
SMS is a developmental disorder that affects about 1 in 15,000–25,000 individuals. It was first described by Ann Smith and Ellen Magenis in the 1980s. It has a specific set of symptoms, including developmental delay, features of autism, sleep disturbances and self-injurious behaviors, among others.
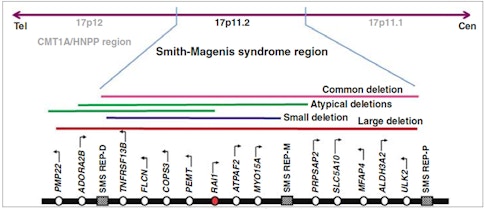
Smith and Magenis teamed up to show that a group of patients that shared these symptoms had the same chromosomal aberration — a microdeletion at chromosomal region 17p11.2 — that deleted several dozen genes. For a long time, people did not know which genes were causal. And then a major discovery was made by Sarah Elsea in 20032, showing that deletion or mutations in one of these genes, RAI1, is responsible for almost all of the symptoms of SMS. So that was a very important discovery, which helped people to focus their efforts on the RAI1 gene.
Approximately 10 percent of individuals with SMS have deletions or mutations in the RAI1 gene. What is RAI1 and what was previously known about how dysfunction of this gene contributes to SMS symptoms?
RAI1 was originally identified in cell culture studies as a gene that’s induced by retinoic acids — the number one Retinoic Acid Induced gene, so RAI1. However, there is so far very little indication that RAI1 is relevant to retinoic acid signaling in other systems. So the name itself does not mean much. What we do know is that RAI1 is a large gene that is highly conserved from mouse to human, and it is a nuclear protein that has two recognizable domains that can potentially bind chromatin. Previous studies have shown that RAI1 could bind to chromatin in vitro, and over-expression studies of this gene suggested that it could cause some transcriptional activation.
Then, in 2007, James Lupski’s lab at Baylor College of Medicine made a mouse model that lacks RAI1 globally. They found that heterozygous Rai1 mice mimicked certain aspects of SMS, such as the facial cranial abnormalities seen in humans3. But the neurodevelopmental features were a little more difficult to understand, as heterozygous Rai1 mice seemed to have a very mild neural phenotype. The majority of Rai1 homozygous mice die in embryogenesis — actually before nervous system development. So Rai1 clearly has pleiotropic effects on the development of many tissues.
“Studying syndromic disorders, like SMS and Rett syndrome — which share features of autism but are caused by a single gene with a clear etiology and molecular function — allows us to find commonalities and differences and will pave the way for future studies of many more genes that affect autism.”
Your lab recently published a paper, supported by SFARI funding, showing that RAI1 has particular roles in distinct neuronal cell types in mice. What do these findings tell us about how RAI1 causes SMS symptoms?
There are multiple models that, for complex neurodevelopmental disorders, could explain how disruption of a particular gene can cause various different phenotypes. One possibility is that Rai1 is required in a particular cell type that’s responsible for one phenotype — cell type A is responsible for phenotype A, cell type B is responsible for phenotype B, and so on. Another possibility is that there is only one major cell type that’s important, and it accounts for all the phenotypes. And the third possibility is actually more complex, with the extreme possibility being that every cell type contributes to every phenotype. Our recent study helped us to understand how different neuronal cell types contribute to SMS phenotypes1.
First of all, we made a conditional Rai1 knockout mouse that removed Rai1 from all neurons without causing early lethality, so we could assay various different phenotypes. We found that these mice had a series of phenotypes, including alterations in motor function, learning deficits and obesity.
Second, we generated mouse lines that had cell type–specific knockout of Rai1 in distinct neuronal cell types — from very broad removal, such as in all inhibitory neurons or in all cortical or subcortical excitatory neurons, to removal in specific neuronal subtypes. And our general take-home message is — it’s complex. That is, certain phenotypes are caused by disruption of Rai1 in multiple cell types and other cell types contribute less. And for some phenotypes, we can ascribe one particular cell type as contributing. So this kind of systematic analysis indicates the complexity of this neurodevelopmental disorder and helps us to understand how distinct neuronal cell types contribute to SMS phenotypes.
You looked at motor and learning abilities and obesity in these mice, three of the main phenotypes that individuals with SMS exhibit. Could you tell us more about those findings?
Yes. Obesity is the simplest phenotype because we know a lot about the neural control of eating and it is relatively easy to study it objectively in mice. In Rai1 conditional knockout mice, the obesity effect is dosage sensitive, which sort of mimics the human condition. In our study, we were actually able to show that loss of RAI1 in a few specific nuclei in the hypothalamus is likely to be the main contributor to the obesity phenotype.
Liqun Luo Laboratory/Stanford University
Of course, what people would foremost like to treat are the cognitive phenotypes. But we are still limited by what a mouse can do. So while learning deficits are obviously the ones that we would like to attack the most, it’s also one of the most difficult because many systems can affect learning. Furthermore, both global and conditional Rai1 heterozygous mice do not exhibit learning deficits with the standard assays so far.
Another major problem in individuals with SMS is behavioral control — impulsivity. And there are not very many good assays to assess the impulsivity of a mouse. So currently, we are continuing studies to test more behavioral defects, including identifying better assays that mimic the symptoms we most want to treat in humans.
We also carried out molecular genomic experiments as part of this study. Because Rai1 is a transcription regulator, we identified transcriptional targets regulated by the Rai1 protein. One of the most exciting targets that we identified in the hypothalamus was 5-HT2C — a serotonin receptor. Our hope is to integrate these kind of molecular approaches and neural, cell type–specific approaches to home in on distinct mechanisms that affect different behavioral aspects of SMS.
How do you see your findings impacting the development of potential treatment strategies for SMS?
So it’s a long road to treatment, as is the case for many neurodevelopmental disorders. One possibility is that it will help clinicians to focus on specific cell types that are the most important for the symptoms that are most limiting for a particular individual. In the future — and I’m not saying anytime soon, but someday — we hope to be able to use gene-therapy approaches to replace a mutated copy of a gene with a wild-type copy. For example, with CRISPR/Cas9 technology, we can very precisely correct genes (at least in animal models). Eventually, if and when we have safe viral vectors that allow us to perform gene editing in specific types of neurons, then this type of work (i.e., identifying the specific cells that are the most critical for particular phenotypes) will become very important.
For a neurodevelopmental disorder such as SMS, there is another caveat to gene therapy (or, indeed, any therapeutic approach) and that is whether you can correct a neurodevelopmental defect in the adult. We don’t yet know whether this is possible for RAI1. But in the case of Rett syndrome — another neurodevelopmental disorder where the causal gene (MECP2) is required throughout development — some of the symptoms were able to be alleviated in a mouse model by delayed restoration of the gene, including in adult mice4. So while it’s a developmental disorder, it is not irreversible, at least in animal models.
You recently received additional SFARI funding to continue your work on SMS and RAI1. What do you think are the most critical outstanding questions and what next steps is your lab planning to take?
Yes. As I mentioned, especially when we talk about higher cognitive functions, the mouse model is limited. A lot of times, the mouse models do not completely mimic the symptoms of the human condition. For instance, Rai1 heterozygous mice don’t have “comparable symptoms” to SMS. I think there are two main reasons for this. One is that the physiology of the human brain and the mouse brain can be quite different. After all, we have many more neurons and much more sophisticated cognitive functions. And the second is the limitations of the existing behavioral assays to have face validity with the human conditions we are studying. We are trying to develop more phenotypic assays in collaboration with our behavioral core. And I hope researchers will continue to work hard to try to improve and enrich behavioral assays in rodent models. Once we better mimic the human symptoms, we can then use the mouse for therapeutic intervention testing as well.
I also alluded earlier to one of the major questions for neurodevelopmental disorders — whether you can rescue some of the SMS phenotypes by introducing RAI1 back into an adult nervous system. With support from our SFARI grant, we are testing the temporal requirement of Rai1. We have a conditional rescue strategy that’s tamoxifen inducible, and we’re doing experiments to induce Rai1 expression at different developmental stages or in adulthood, to see whether we can rescue any of the behavioral phenotypes in the mouse model. We are also starting to delve deeper into physiology — we have obvious molecular and behavioral alterations in these mice, but what’s wrong with neurons in the circuits? This work will help bridge the gap between the molecular and behavioral studies.
You, and many other researchers, are making great strides in understanding the biological causes of autism and related neurodevelopmental disorders. What about these scientific discoveries gives you the most hope?
Autism is a very heterogeneous neurodevelopmental disorder and has many different contributing factors, and we are still identifying more and more genes that contribute to autism. I think studying syndromic disorders, like SMS and Rett syndrome — which share features of autism but are caused by a single gene with a clear etiology and molecular function — allows us to find commonalities and differences and will pave the way for future studies of many more genes that affect autism.
One of the worst-case scenarios in neurodevelopmental disorders would be that something happens early on in development that is irreversible and that nothing could be done later on to ameliorate the disorder’s symptoms. So of all the recent studies on neurodevelopmental disorders, some of the most exciting findings to me are the studies that show that, if you restore a gene in the adult animal, you can correct many of the symptoms4,5. That in itself does not provide a specific therapeutic strategy. But it suggests that once you find a strategy, it may not be too late to treat. So those rescue experiments in mice, I feel, are the most inspiring experiments in neurodevelopmental disorders.
I noticed that you dedicated your recent work to your daughter and other individuals with SMS. Is there any message that you, as a parent advocate, would like to send to the scientific community?
As I mentioned at a recent SFARI meeting, when you compare SMS to Rett syndrome, they both have prevalence rates on the same order of magnitude, and the disorders are caused by mutations in similar kinds of proteins. RAI1 and MeCP2 are both nuclear factors that regulate chromatin. But the number of scientists who study Rett syndrome is magnitudes more than the number that studies SMS. So I would like to make an appeal to researchers to pursue further studies of SMS. I’ll be happy to contribute, if researchers want mice, antibodies.
I also want to mention how very appreciative I am of two talented people in my lab — Wei-Hsiang Huang, a postdoctoral fellow, and Casey Guenthner, a graduate student, the two first co-authors on the RAI1 paper1. My lab is not a developmental disorder lab, and without them, I wouldn’t be studying this disorder, even though I have a personal interest in this, so I’m very appreciative to both of these scientists.
References
- Huang W.H., Guenthner C.J., Xu J., Nguyen T., Schwarz L.A., Wilkinson A.W., Gozani O., Chang H.Y., Shamloo M., Luo L. Molecular and neural functions of Rai1, the causal gene for Smith-Magenis syndrome. Neuron 92, 392–406 (2016) PubMed
- Slager R.E., Newton T.L., Vlangos C.N., Finucane B., Elsea S.H. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat. Genet. 33, 466–468 (2003) PubMed
- Bi W., Yan J., Shi X., Yuva-Paylor L.A., Antalffy B.A., Goldman A., Yoo J.W., Noebels J.L., Armstrong D.L., Paylor R., Lupski J.R. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum. Mol. Genet. 16, 1802–1813 (2007) PubMed
- Guy J., Gan J., Selfridge J., Cobb S., Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147 (2007) PubMed
- Ehninger D., Li W., Fox K., Stryker M.P., Silva A.J. Reversing neurodevelopmental disorders in adults. Neuron 60, 950–960 (2008) PubMed
- Elsea S.H. and S. Girirajan Smith-Magenis syndrome. Eur. J. Hum. Genet. 16, 412–421 (2008) PubMed


